

Explicable prioritization of genetic variants by integration of rule-based and machine learning algorithms for diagnosis of rare Mendelian disorders
- PMID: 38509596
- PMCID: PMC10956189
- DOI: 10.1186/s40246-024-00595-8
Explicable prioritization of genetic variants by integration of rule-based and machine learning algorithms for diagnosis of rare Mendelian disorders
Abstract
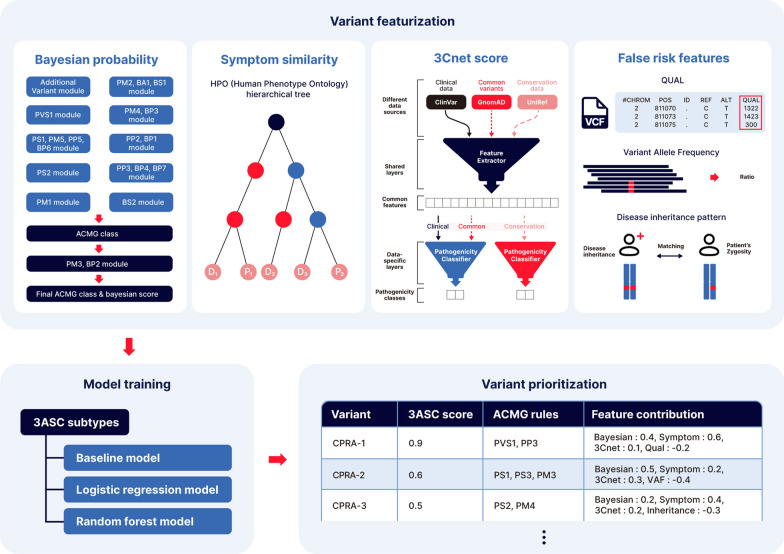
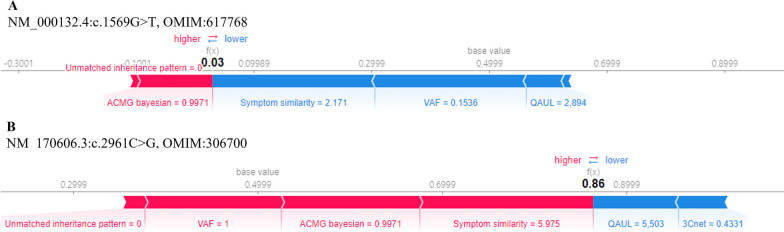
Background: In the process of finding the causative variant of rare diseases, accurate assessment and prioritization of genetic variants is essential. Previous variant prioritization tools mainly depend on the in-silico prediction of the pathogenicity of variants, which results in low sensitivity and difficulty in interpreting the prioritization result. In this study, we propose an explainable algorithm for variant prioritization, named 3ASC, with higher sensitivity and ability to annotate evidence used for prioritization. 3ASC annotates each variant with the 28 criteria defined by the ACMG/AMP genome interpretation guidelines and features related to the clinical interpretation of the variants. The system can explain the result based on annotated evidence and feature contributions.
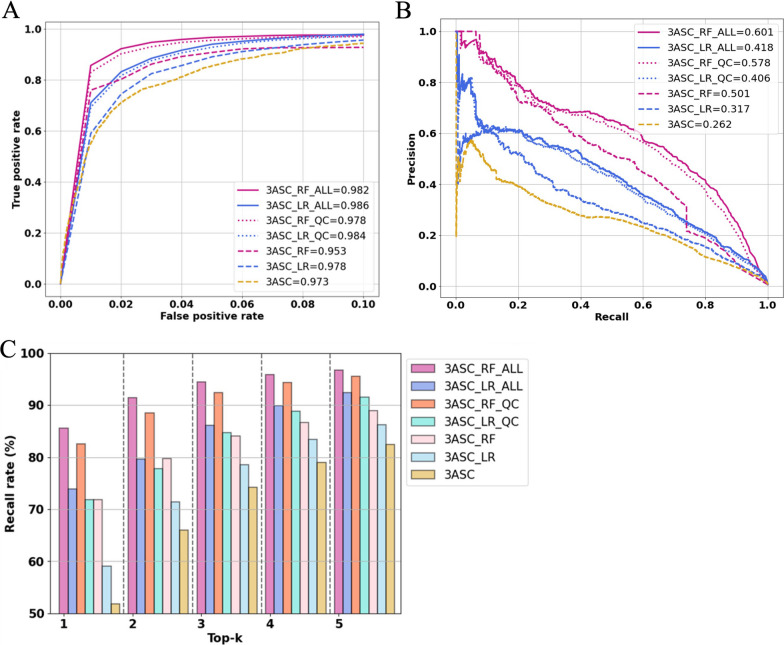
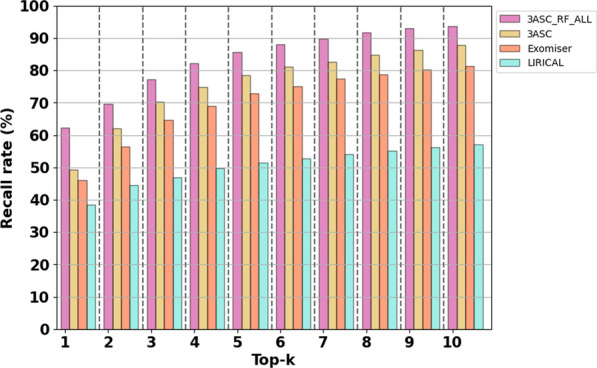
Results: We trained various machine learning algorithms using in-house patient data. The performance of variant ranking was assessed using the recall rate of identifying causative variants in the top-ranked variants. The best practice model was a random forest classifier that showed top 1 recall of 85.6% and top 3 recall of 94.4%. The 3ASC annotates the ACMG/AMP criteria for each genetic variant of a patient so that clinical geneticists can interpret the result as in the CAGI6 SickKids challenge. In the challenge, 3ASC identified causal genes for 10 out of 14 patient cases, with evidence of decreased gene expression for 6 cases. Among them, two genes (HDAC8 and CASK) had decreased gene expression profiles confirmed by transcriptome data.
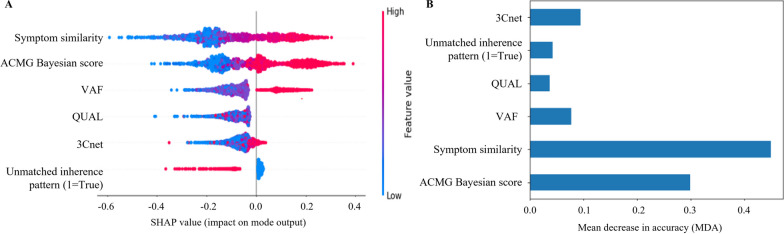
Conclusions: 3ASC can prioritize genetic variants with higher sensitivity compared to previous methods by integrating various features related to clinical interpretation, including features related to false positive risk such as quality control and disease inheritance pattern. The system allows interpretation of each variant based on the ACMG/AMP criteria and feature contribution assessed using explainable AI techniques.
Keywords: Clinical genome interpretation; Explainable AI; Mendelian disorder; Variant prioritization.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Kim HH, Woo J, Kim D-W, Lee J, Seo GH, Lee H, et al. Disease-causing variant recommendation system for clinical genome interpretation with adjusted scores for artefactual variants. bioRxiv [Internet]. 2022; Available from: https://www.biorxiv.org/content/early/2022/10/14/2022.10.12.511857
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
