

VIPERA: Viral Intra-Patient Evolution Reporting and Analysis
- PMID: 38510921
- PMCID: PMC10953798
- DOI: 10.1093/ve/veae018
VIPERA: Viral Intra-Patient Evolution Reporting and Analysis
Abstract
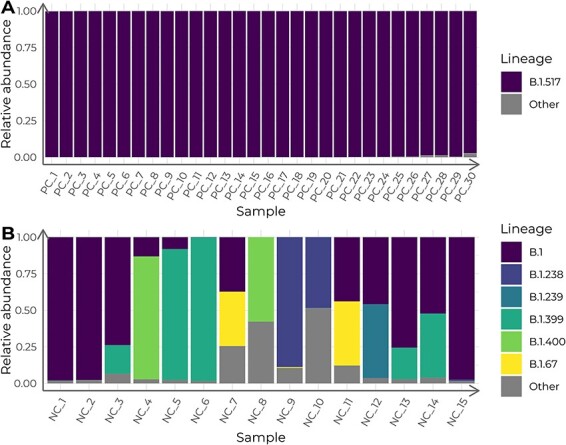
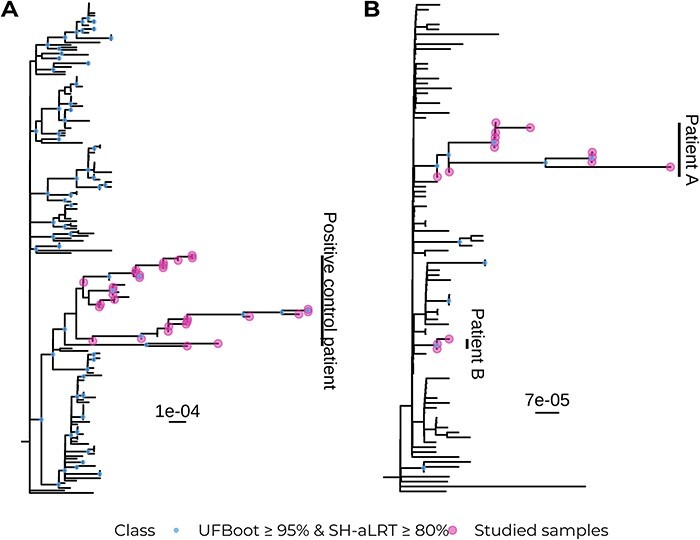
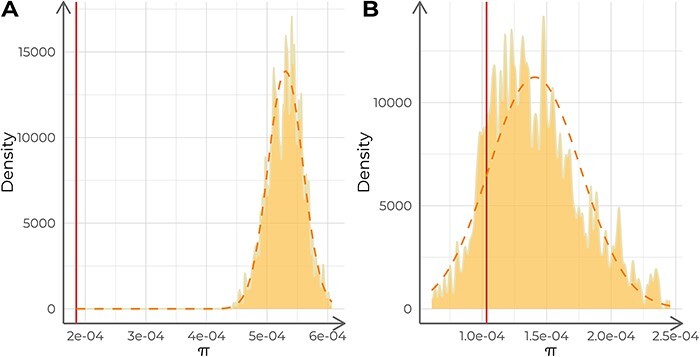
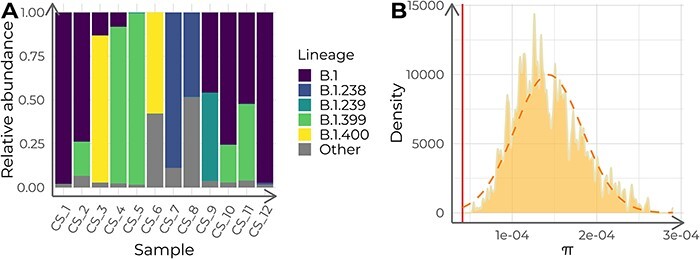
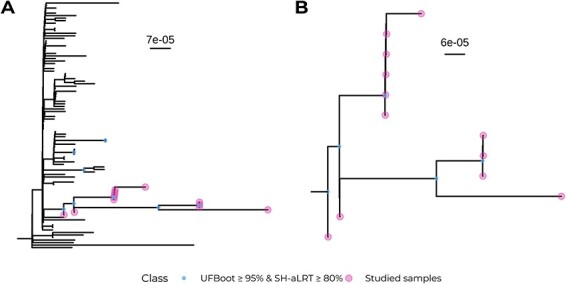
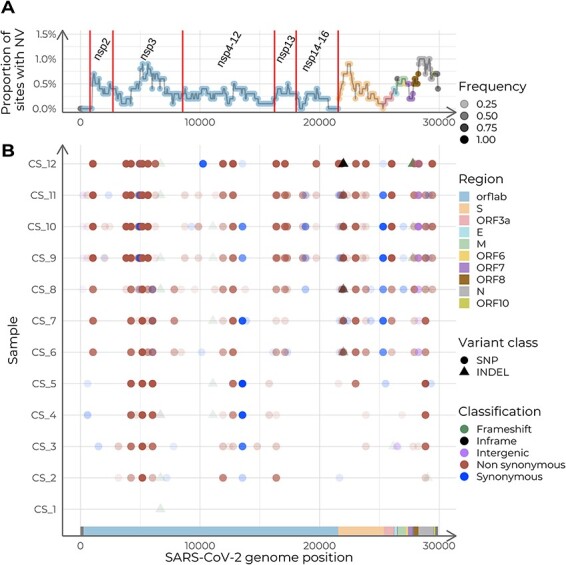
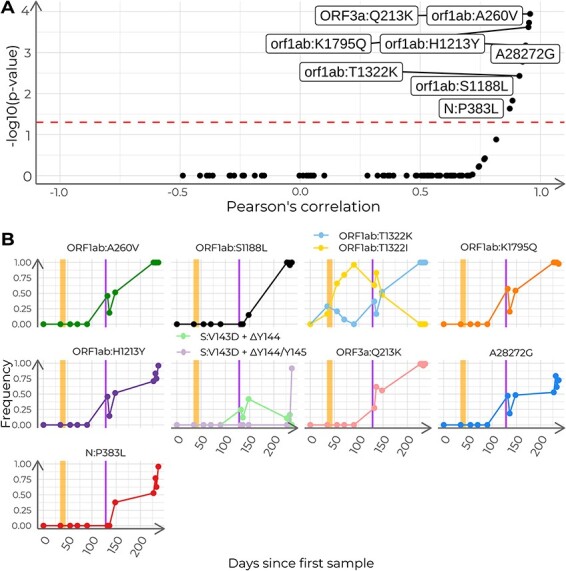
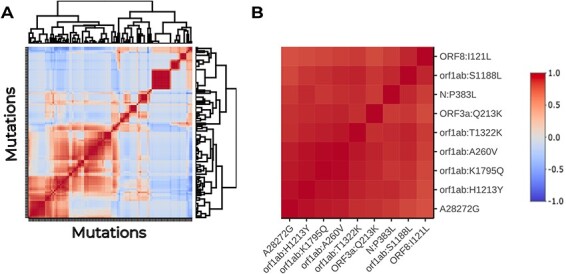
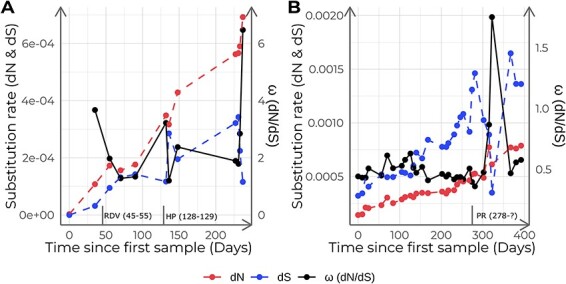
Viral mutations within patients nurture the adaptive potential of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) during chronic infections, which are a potential source of variants of concern. However, there is no integrated framework for the evolutionary analysis of intra-patient SARS-CoV-2 serial samples. Herein, we describe Viral Intra-Patient Evolution Reporting and Analysis (VIPERA), a new software that integrates the evaluation of the intra-patient ancestry of SARS-CoV-2 sequences with the analysis of evolutionary trajectories of serial sequences from the same viral infection. We have validated it using positive and negative control datasets and have successfully applied it to a new case, which revealed population dynamics and evidence of adaptive evolution. VIPERA is available under a free software license at https://github.com/PathoGenOmics-Lab/VIPERA.
Keywords: SARS-CoV-2; bioinformatics; intra-patient diversity; serially sampled infection; snakemake workflow; within-host evolution.
© The Author(s) 2024. Published by Oxford University Press.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Andersen Laboratory . (2023) Freyja: Depth-weighted De-Mixing. <https://github.com/andersen-lab/Freyja> accessed 16 Jun 2023.
-
- ARTICnetwork . (2023) ARTIC-ncov2019: ARTIC Nanopore Protocol for nCoV2019 Novel Coronavirus. <https://github.com/artic-network/artic-ncov2019> accessed 7 Jun 2023.
-
- Benjamini Y., and Hochberg Y. (1995) ‘Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing’, Journal of the Royal Statistical Society: Series B (Methodological), 57: 289–300.
LinkOut - more resources
Full Text Sources
Miscellaneous
