

RUNX1 C-terminal mutations impair blood cell differentiation by perturbing specific enhancer-promoter networks
- PMID: 38513139
- PMCID: PMC11112616
- DOI: 10.1182/bloodadvances.2023011484
RUNX1 C-terminal mutations impair blood cell differentiation by perturbing specific enhancer-promoter networks
Abstract
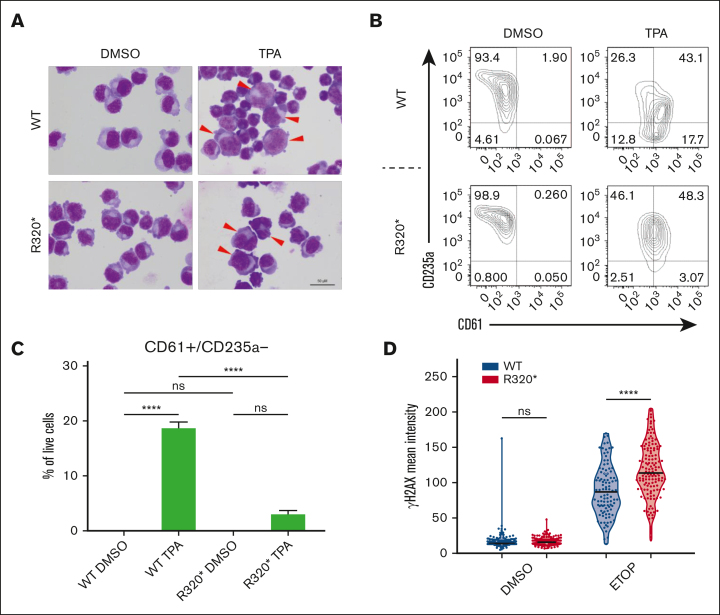
The transcription factor RUNX1 is a master regulator of hematopoiesis and is frequently mutated in myeloid malignancies. Mutations in its runt homology domain (RHD) frequently disrupt DNA binding and result in loss of RUNX1 function. However, it is not clearly understood how other RUNX1 mutations contribute to disease development. Here, we characterized RUNX1 mutations outside of the RHD. Our analysis of the patient data sets revealed that mutations within the C-terminus frequently occur in hematopoietic disorders. Remarkably, most of these mutations were nonsense or frameshift mutations and were predicted to be exempt from nonsense-mediated messenger RNA decay. Therefore, this class of mutation is projected to produce DNA-binding proteins that contribute to the pathogenesis in a distinct manner. To model this, we introduced the RUNX1R320∗ mutation into the endogenous gene locus and demonstrated the production of RUNX1R320∗ protein. Expression of RUNX1R320∗ resulted in the disruption of RUNX1 regulated processes such as megakaryocytic differentiation, through a transcriptional signature different from RUNX1 depletion. To understand the underlying mechanisms, we used Global RNA Interactions with DNA by deep sequencing (GRID-seq) to examine enhancer-promoter connections. We identified widespread alterations in the enhancer-promoter networks within RUNX1 mutant cells. Additionally, we uncovered enrichment of RUNX1R320∗ and FOXK2 binding at the MYC super enhancer locus, significantly upregulating MYC transcription and signaling pathways. Together, our study demonstrated that most RUNX1 mutations outside the DNA-binding domain are not subject to nonsense-mediated decay, producing protein products that act in concert with additional cofactors to dysregulate hematopoiesis through mechanisms distinct from those induced by RUNX1 depletion.
© 2024 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: B.R. is a cofounder of Epigenome Technologies, Inc and has equity in Arima Genomics, Inc. The remaining authors declare no competing financial interests.
The current affiliation for Z.L. is School of Life Sciences, Southern University of Science and Technology, Shenzhen, Guangdong, China.
The current affiliation for D.-H.L. is School of Systems Biomedical Science, Soongsil University, Seoul, Republic of Korea.
The current affiliation for P.B.C. is Genome Institute of Singapore, Agency for Science, Technology and Research (A∗STAR), Singapore.
Figures






References
-
- Okuda T, Van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84(2):321–330. - PubMed
