

Diversity of CFTR variants across ancestries characterized using 454,727 UK biobank whole exome sequences
- PMID: 38515211
- PMCID: PMC10956269
- DOI: 10.1186/s13073-024-01316-5
Diversity of CFTR variants across ancestries characterized using 454,727 UK biobank whole exome sequences
Abstract
Background: Limited understanding of the diversity of variants in the cystic fibrosis transmembrane conductance regulator (CFTR) gene across ancestries hampers efforts to advance molecular diagnosis of cystic fibrosis (CF). The consequences pose a risk of delayed diagnoses and subsequently worsened health outcomes for patients. Therefore, characterizing the spectrum of CFTR variants across ancestries is critical for revolutionizing molecular diagnoses of CF.
Methods: We analyzed 454,727 UK Biobank (UKBB) whole-exome sequences to characterize the diversity of CFTR variants across ancestries. Using the PanUKBB classification, the participants were assigned into six major groups: African (AFR), American/American Admixed (AMR), Central South Asia (CSA), East Asian (EAS), European (EUR), and Middle East (MID). We segregated ancestry-specific CFTR variants, including those that are CF-causing or clinically relevant. The ages of certain CF-causing variants were determined and analyzed for selective pressure effects, and curated phenotype analysis was performed for participants with clinically relevant CFTR genotypes.
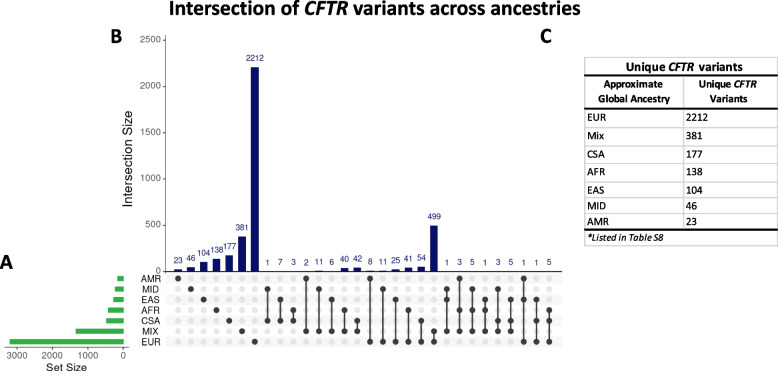
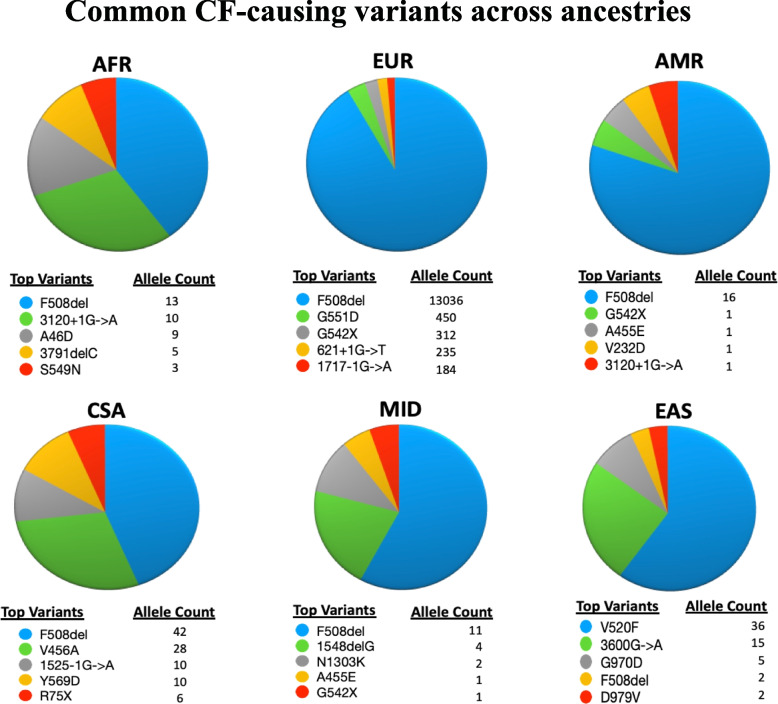
Results: We detected over 4000 CFTR variants, including novel ancestry-specific variants, across six ancestries. Europeans had the most unique CFTR variants [n = 2212], while the American group had the least unique variants [n = 23]. F508del was the most prevalent CF-causing variant found in all ancestries, except in EAS, where V520F was the most prevalent. Common EAS variants such as 3600G > A, V456A, and V520, which appeared approximately 270, 215, and 338 generations ago, respectively, did not show evidence of selective pressure. Sixteen participants had two CF-causing variants, with two being diagnosed with CF. We found 154 participants harboring a CF-causing and varying clinical consequences (VCC) variant. Phenotype analysis performed for participants with multiple clinically relevant variants returned significant associations with CF and its pulmonary phenotypes [Bonferroni-adjusted p < 0.05].
Conclusions: We leveraged the UKBB database to comprehensively characterize the broad spectrum of CFTR variants across ancestries. The detection of over 4000 CFTR variants, including several ancestry-specific and uncharacterized CFTR variants, warrants the need for further characterization of their functional and clinical relevance. Overall, the presentation of classical CF phenotypes seen in non-CF diagnosed participants with more than one CF-causing variant indicates that they may benefit from current CFTR modulator therapies.
Keywords: CFTR; Cystic fibrosis; UK biobank; Whole exome sequencing.
© 2024. The Author(s).
Conflict of interest statement
JEI, ML, BRG, SRP, FR, PK, RT, PD, AS, JFW, and AV are employees of AbbVie. All authors declare that they have no competing interests.
Figures



Similar articles
-
Corrector therapies (with or without potentiators) for people with cystic fibrosis with class II CFTR gene variants (most commonly F508del).Cochrane Database Syst Rev. 2020 Dec 17;12(12):CD010966. doi: 10.1002/14651858.CD010966.pub3. Cochrane Database Syst Rev. 2020. Update in: Cochrane Database Syst Rev. 2023 Nov 20;11:CD010966. doi: 10.1002/14651858.CD010966.pub4. PMID: 33331662 Free PMC article. Updated.
-
Complete CFTR gene sequencing in 5,058 individuals with cystic fibrosis informs variant-specific treatment.J Cyst Fibros. 2022 May;21(3):463-470. doi: 10.1016/j.jcf.2021.10.011. Epub 2021 Nov 12. J Cyst Fibros. 2022. PMID: 34782259
-
Mutational spectrum of CFTR in cystic fibrosis patients with gastrointestinal and hepatobiliary manifestations.Mol Biol Rep. 2024 Apr 25;51(1):573. doi: 10.1007/s11033-024-09508-3. Mol Biol Rep. 2024. PMID: 38662334
-
DNA sequencing analysis of cystic fibrosis transmembrane conductance regulator gene identifies cystic fibrosis-associated variants in the Severe Asthma Research Program.Pediatr Pulmonol. 2022 Jul;57(7):1782-1788. doi: 10.1002/ppul.25939. Epub 2022 May 5. Pediatr Pulmonol. 2022. PMID: 35451201 Free PMC article.
-
Exploring the diversity of CFTR gene mutations in cystic fibrosis individuals of South Asia.J Asthma. 2024 Jun;61(6):511-519. doi: 10.1080/02770903.2023.2297365. Epub 2023 Dec 28. J Asthma. 2024. PMID: 38153325 Review.
Cited by
-
Identified five variants in CFTR gene that alter RNA splicing by minigene assay.Front Genet. 2025 Mar 20;16:1543623. doi: 10.3389/fgene.2025.1543623. eCollection 2025. Front Genet. 2025. PMID: 40182926 Free PMC article.
-
The Spectrum and Carrier Frequencies of Common Pathogenic Cystic Fibrosis Transmembrane Conductance Regulator Gene Mutations in Men from the General Population: The Role of Ethnicity.Int J Mol Sci. 2025 Jul 10;26(14):6625. doi: 10.3390/ijms26146625. Int J Mol Sci. 2025. PMID: 40724872 Free PMC article.
-
[Detection of pathogenic gene mutations in thirteen cases of congenital bilateral absence of vas deferens infertility patients].Beijing Da Xue Xue Bao Yi Xue Ban. 2024 Oct 18;56(5):763-774. doi: 10.19723/j.issn.1671-167X.2024.05.003. Beijing Da Xue Xue Bao Yi Xue Ban. 2024. PMID: 39397452 Free PMC article. Chinese.
-
Refining CFTR-Related Metabolic Syndrome (CRMS)/Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID) Diagnosis: Impact of CFTR2 Variant Classifications.Int J Neonatal Screen. 2025 Jul 30;11(3):60. doi: 10.3390/ijns11030060. Int J Neonatal Screen. 2025. PMID: 40843902 Free PMC article. Review.
-
Elexacaftor/tezacaftor/ivacaftor in children aged ≥6 years with cystic fibrosis heterozygous for F508del and a minimal function mutation: results from a 96-week open-label extension study.Eur Respir J. 2025 Jul 14;66(1):2402435. doi: 10.1183/13993003.02435-2024. Print 2025 Jul. Eur Respir J. 2025. PMID: 40210412 Free PMC article. Clinical Trial.
References
-
- Griese M, et al. Safety and Efficacy of Elexacaftor/Tezacaftor/Ivacaftor for 24 Weeks or Longer in People with Cystic Fibrosis and One or More F508del Alleles: Interim Results of an Open-Label Phase 3 Clinical Trial. Am J Respir Crit Care Med. 2021;203:381–385. doi: 10.1164/rccm.202008-3176LE. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
