

BET inhibitors drive Natural Killer activation in non-small cell lung cancer via BRD4 and SMAD3
- PMID: 38519469
- PMCID: PMC10960013
- DOI: 10.1038/s41467-024-46778-8
BET inhibitors drive Natural Killer activation in non-small cell lung cancer via BRD4 and SMAD3
Abstract
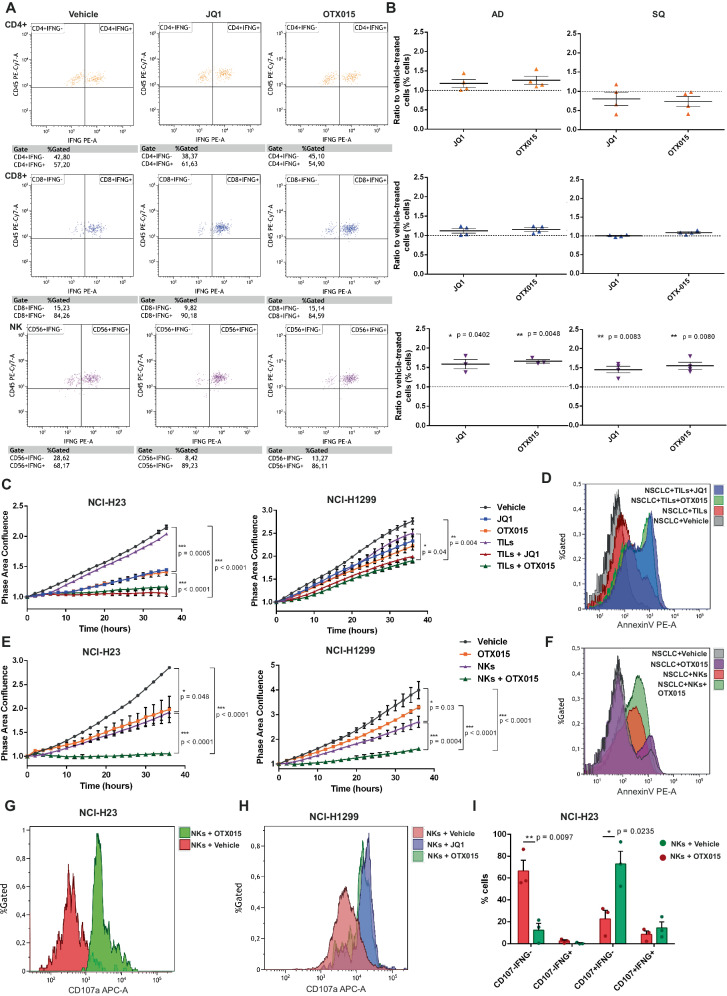
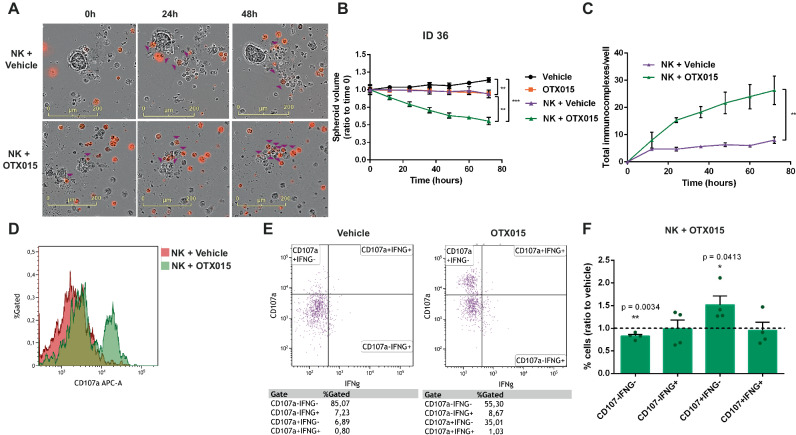
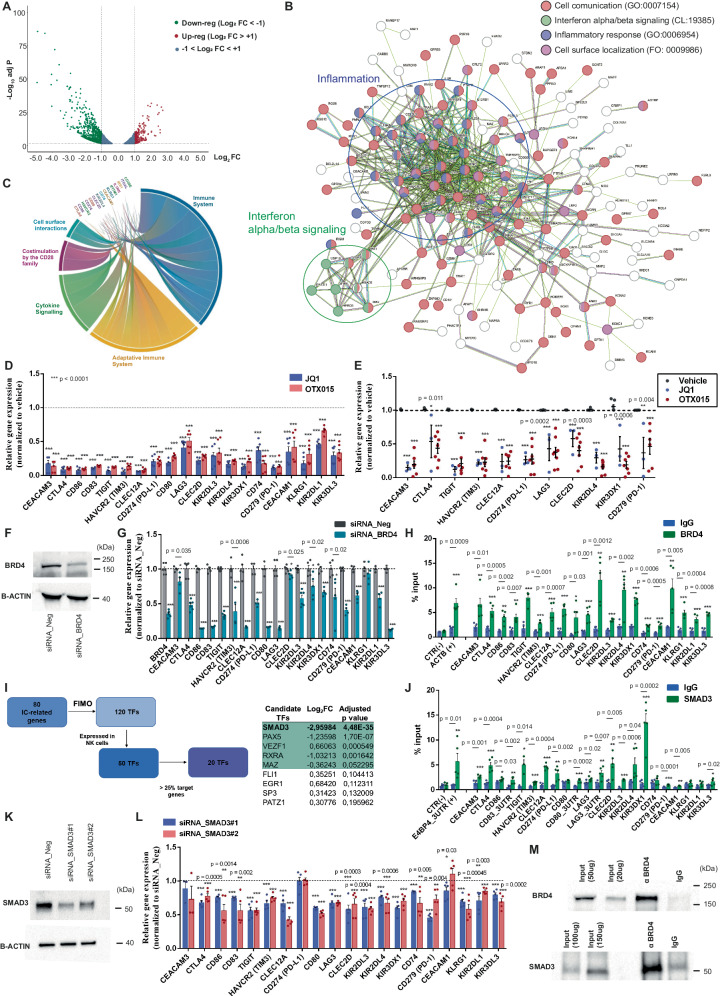
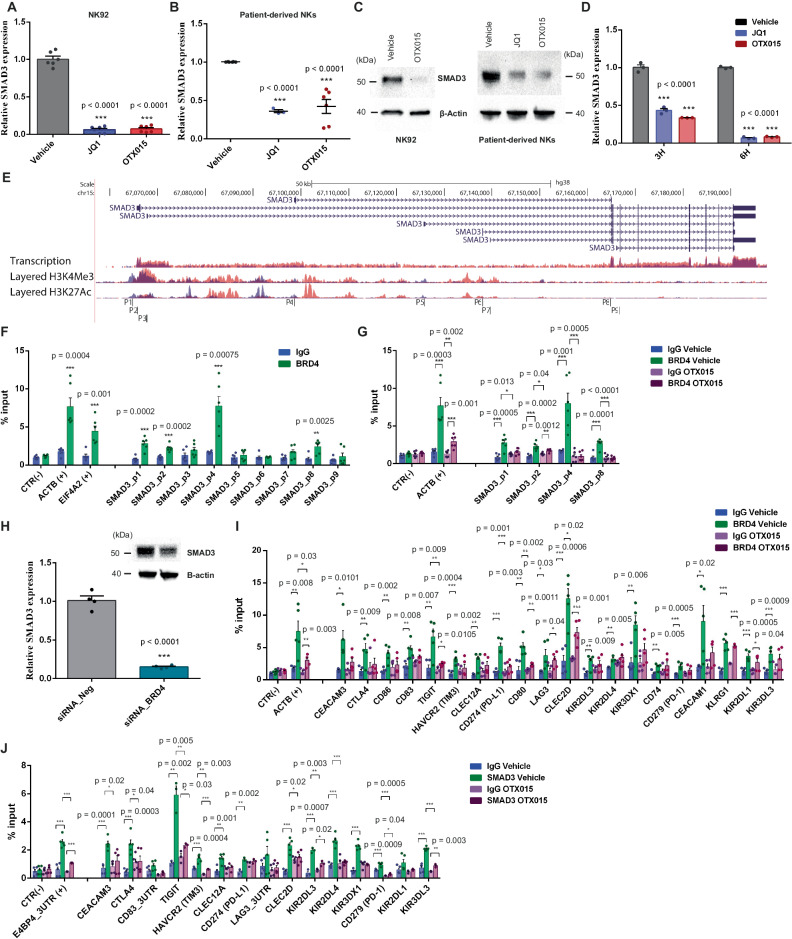
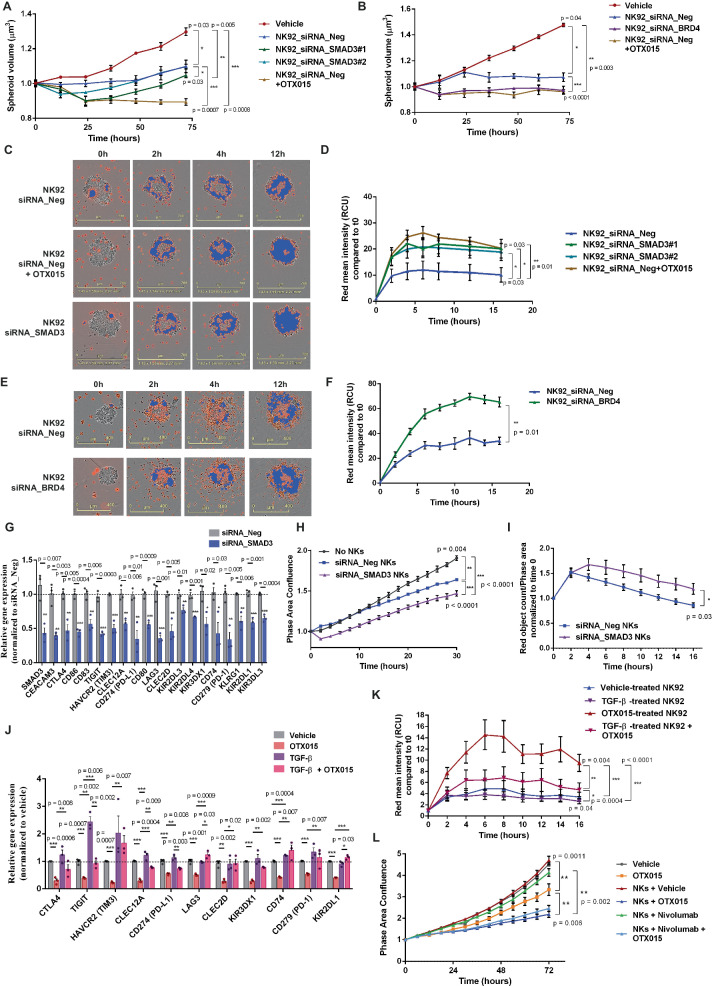
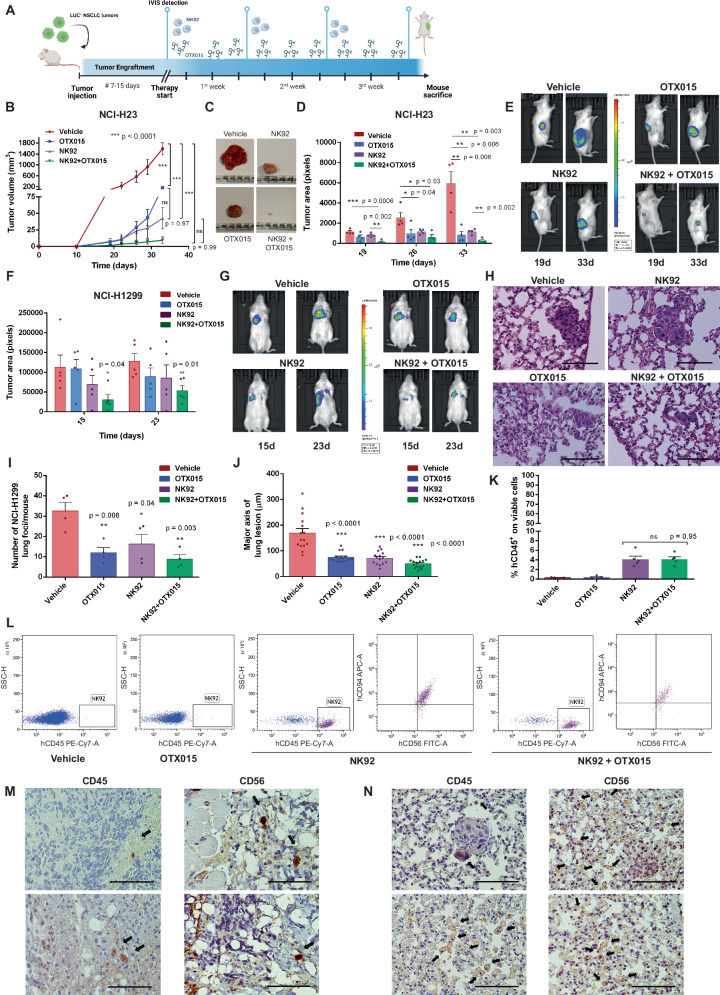
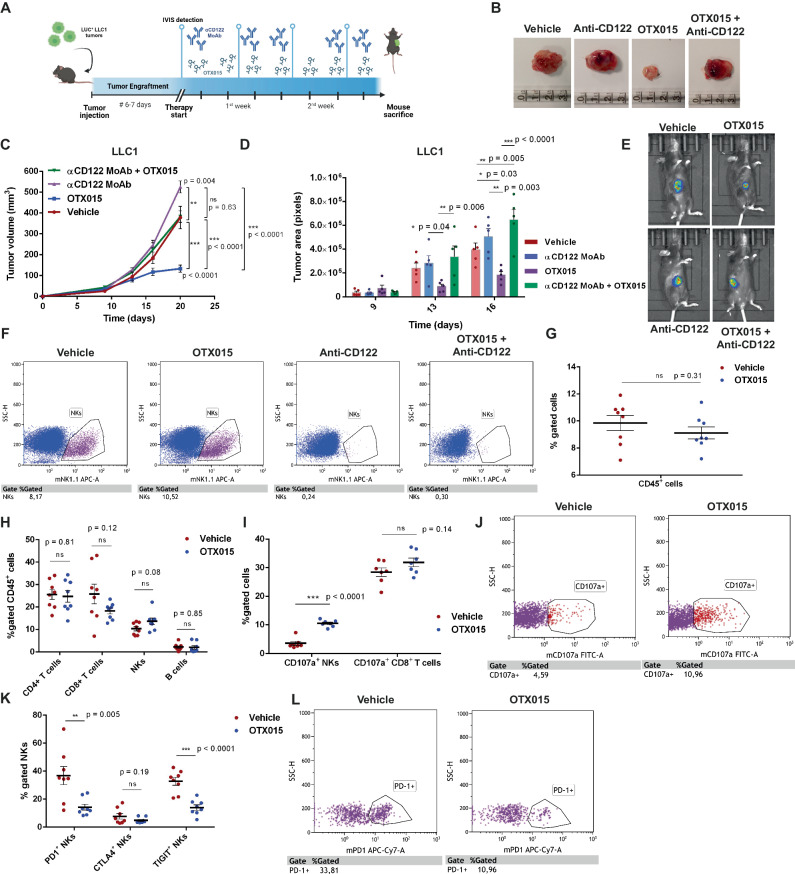
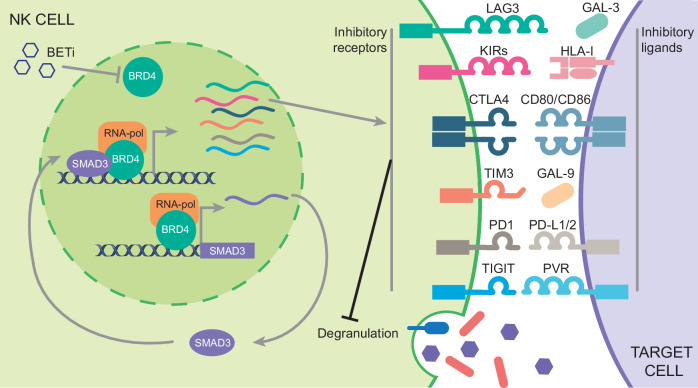
Non-small-cell lung carcinoma (NSCLC) is the most common lung cancer and one of the pioneer tumors in which immunotherapy has radically changed patients' outcomes. However, several issues are emerging and their implementation is required to optimize immunotherapy-based protocols. In this work, we investigate the ability of the Bromodomain and Extra-Terminal protein inhibitors (BETi) to stimulate a proficient anti-tumor immune response toward NSCLC. By using in vitro, ex-vivo, and in vivo models, we demonstrate that these epigenetic drugs specifically enhance Natural Killer (NK) cell cytotoxicity. BETi down-regulate a large set of NK inhibitory receptors, including several immune checkpoints (ICs), that are direct targets of the transcriptional cooperation between the BET protein BRD4 and the transcription factor SMAD3. Overall, BETi orchestrate an epigenetic reprogramming that leads to increased recognition of tumor cells and the killing ability of NK cells. Our results unveil the opportunity to exploit and repurpose these drugs in combination with immunotherapy.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
- Bando per la Valorizzazione della Ricerca Istituzionale 2021 - Fondi 5 per Mille 2016/Ministero della Salute (Ministry of Health, Italy)
- IG26377/Associazione Italiana per la Ricerca sul Cancro (Italian Association for Cancer Research)
- IG20109/Associazione Italiana per la Ricerca sul Cancro (Italian Association for Cancer Research)
- IG21772/Associazione Italiana per la Ricerca sul Cancro (Italian Association for Cancer Research)
- Fellowship/Fondazione Umberto Veronesi (Umberto Veronesi Foundation)
LinkOut - more resources
Full Text Sources
Medical
Research Materials
