

Activation of the insulin receptor by insulin-like growth factor 2
- PMID: 38521788
- PMCID: PMC10960814
- DOI: 10.1038/s41467-024-46990-6
Activation of the insulin receptor by insulin-like growth factor 2
Abstract
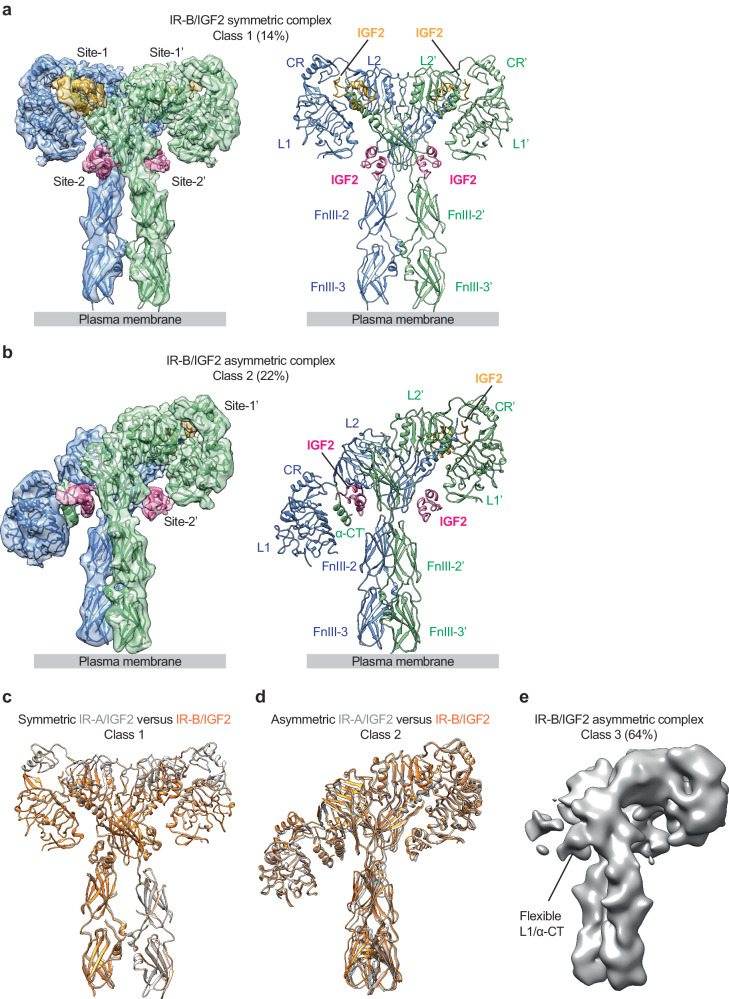
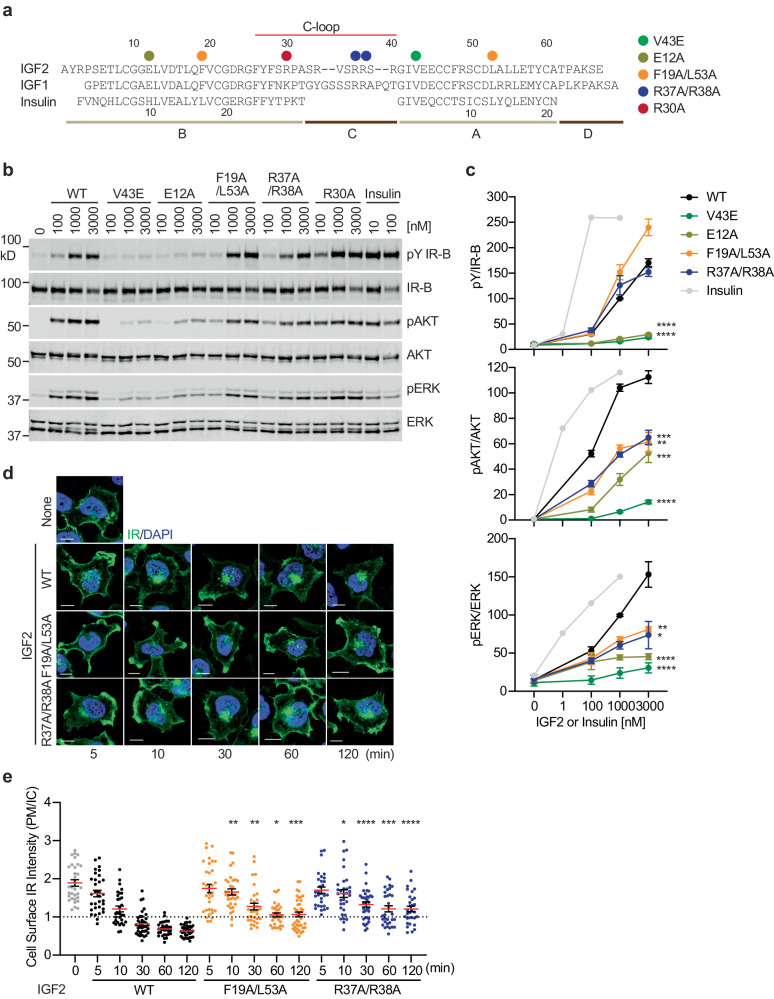
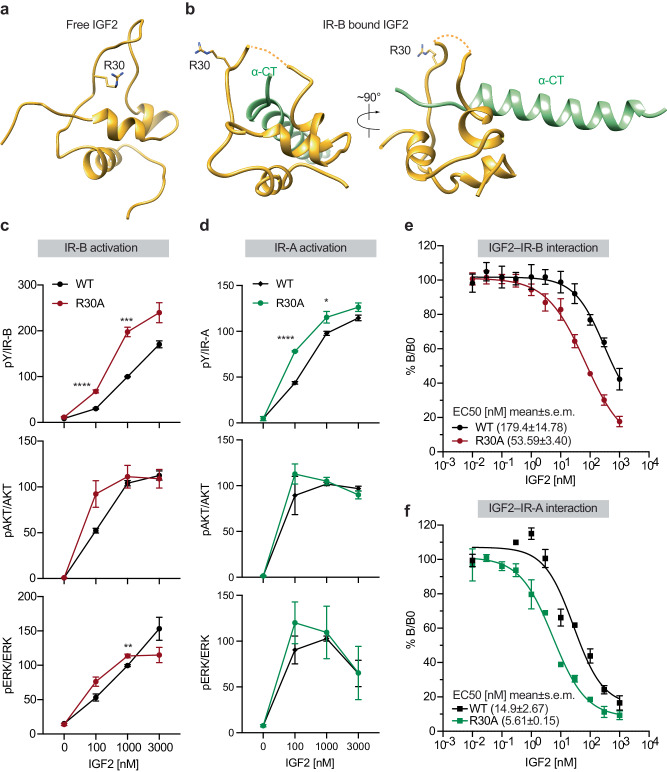
Insulin receptor (IR) controls growth and metabolism. Insulin-like growth factor 2 (IGF2) has different binding properties on two IR isoforms, mimicking insulin's function. However, the molecular mechanism underlying IGF2-induced IR activation remains unclear. Here, we present cryo-EM structures of full-length human long isoform IR (IR-B) in both the inactive and IGF2-bound active states, and short isoform IR (IR-A) in the IGF2-bound active state. Under saturated IGF2 concentrations, both the IR-A and IR-B adopt predominantly asymmetric conformations with two or three IGF2s bound at site-1 and site-2, which differs from that insulin saturated IR forms an exclusively T-shaped symmetric conformation. IGF2 exhibits a relatively weak binding to IR site-2 compared to insulin, making it less potent in promoting full IR activation. Cell-based experiments validated the functional importance of IGF2 binding to two distinct binding sites in optimal IR signaling and trafficking. In the inactive state, the C-terminus of α-CT of IR-B contacts FnIII-2 domain of the same protomer, hindering its threading into the C-loop of IGF2, thus reducing the association rate of IGF2 with IR-B. Collectively, our studies demonstrate the activation mechanism of IR by IGF2 and reveal the molecular basis underlying the different affinity of IGF2 to IR-A and IR-B.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




Similar articles
-
IGF1 and IGF2 specificities to the two insulin receptor isoforms are determined by insulin receptor amino acid 718.PLoS One. 2017 Jun 1;12(6):e0178885. doi: 10.1371/journal.pone.0178885. eCollection 2017. PLoS One. 2017. PMID: 28570711 Free PMC article.
-
Activation mechanism of the insulin receptor revealed by cryo-EM structure of the fully liganded receptor-ligand complex.Elife. 2019 Aug 22;8:e48630. doi: 10.7554/eLife.48630. Elife. 2019. PMID: 31436533 Free PMC article.
-
Effects of Somatostatin Analogs and Dopamine Agonists on Insulin-Like Growth Factor 2-Induced Insulin Receptor Isoform A Activation by Gastroenteropancreatic Neuroendocrine Tumor Cells.Neuroendocrinology. 2016;103(6):815-25. doi: 10.1159/000444280. Epub 2016 Feb 2. Neuroendocrinology. 2016. PMID: 26836610
-
Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease.Endocr Rev. 2009 Oct;30(6):586-623. doi: 10.1210/er.2008-0047. Epub 2009 Sep 14. Endocr Rev. 2009. PMID: 19752219 Review.
-
The Activation Mechanism of the Insulin Receptor: A Structural Perspective.Annu Rev Biochem. 2023 Jun 20;92:247-272. doi: 10.1146/annurev-biochem-052521-033250. Epub 2023 Mar 31. Annu Rev Biochem. 2023. PMID: 37001136 Free PMC article. Review.
Cited by
-
Identifying antisense oligonucleotides for targeted inhibition of insulin receptor isoform A.Front Oncol. 2025 Apr 15;15:1563985. doi: 10.3389/fonc.2025.1563985. eCollection 2025. Front Oncol. 2025. PMID: 40303997 Free PMC article.
-
Structural mechanism of insulin receptor activation by a dimeric aptamer agonist.Exp Mol Med. 2025 Jul;57(7):1506-1518. doi: 10.1038/s12276-025-01494-1. Epub 2025 Jul 2. Exp Mol Med. 2025. PMID: 40603733 Free PMC article.
-
Regulation and function of insulin and insulin-like growth factor receptor signalling.Nat Rev Mol Cell Biol. 2025 Jul;26(7):558-580. doi: 10.1038/s41580-025-00826-3. Epub 2025 Feb 10. Nat Rev Mol Cell Biol. 2025. PMID: 39930003 Review.
-
Paradoxical regulation of IGF2 in promoting lipid metabolism in adipose tissues.Commun Biol. 2025 Jul 9;8(1):1026. doi: 10.1038/s42003-025-08458-1. Commun Biol. 2025. PMID: 40634529 Free PMC article.
-
Insulin-Like Growth Factor Signaling in Alzheimer's Disease: Pathophysiology and Therapeutic Strategies.Mol Neurobiol. 2025 Mar;62(3):3195-3225. doi: 10.1007/s12035-024-04457-1. Epub 2024 Sep 6. Mol Neurobiol. 2025. PMID: 39240280 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
