

Real-World Effectiveness of High-Dose Tafamidis on Neurologic Disease Progression in Mixed-Phenotype Variant Transthyretin Amyloid Cardiomyopathy
- PMID: 38521883
- PMCID: PMC11093936
- DOI: 10.1007/s40119-024-00362-9
Real-World Effectiveness of High-Dose Tafamidis on Neurologic Disease Progression in Mixed-Phenotype Variant Transthyretin Amyloid Cardiomyopathy
Abstract
Introduction: Transthyretin amyloidosis (ATTR) is a progressive, heterogeneous rare disease manifesting as ATTR polyneuropathy (ATTR-PN), ATTR cardiomyopathy (ATTR-CM), or a mixed phenotype. Tafamidis meglumine (20 mg po qd) is approved in some markets to delay neurologic progression in ATTR-PN, while high-dose tafamidis (80/61 mg po qd) is approved worldwide to reduce cardiovascular mortality and cardiovascular-related hospitalization in ATTR-CM. The objective of this study was to assess the real-world benefit of high-dose tafamidis for delaying neurologic progression in patients with mixed-phenotype variant ATTR-CM (ATTRv-CM).
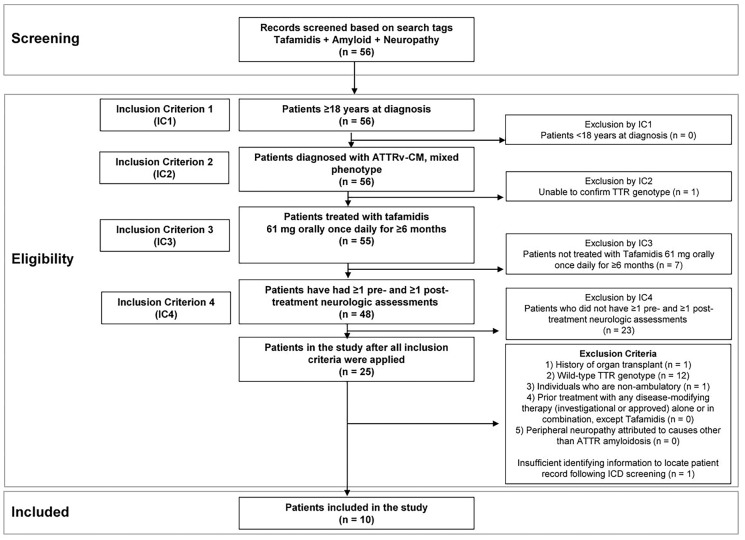
Methods: This exploratory, retrospective, observational cohort study evaluated anonymized electronic medical records and included adult patients with mixed-phenotype ATTRv-CM treated with high-dose tafamidis for at least 6 months. Neurologic assessments included the Medical Research Council (MRC) Scale for Muscle Strength, Neuropathy Impairment Score (NIS) muscle weakness subscale, and Polyneuropathy Disability (PND) instrument. Modified body mass index (mBMI) was also assessed.
Results: Patients (N = 10) started tafamidis treatment an average of 3.8 months after diagnosis, with an average treatment duration of 20.8 months. Seven of 10 patients demonstrated normal muscle strength on the MRC scale throughout the study, and 9 of 10 patients had no decline in muscle strength during the post-treatment period. The NIS muscle weakness subscale score was ≤ 60 for all patients in the study at all time points, suggesting normal function to mild impairment. Six of 10 patients had no change in walking capacity as measured by the PND instrument at pre- and post-assessments, while one-third of patients had a decrease in PND stage (signaling improvement) from pre- to post-assessment. mBMI remained relatively stable throughout the study.
Conclusion: This is the first real-world study to demonstrate the potential value of high-dose tafamidis for delaying neurologic disease progression in patients with mixed-phenotype ATTRv-CM. The findings underscore the importance of multidisciplinary assessment for patients with ATTR amyloidosis.
Trial registration: ClinicalTrials.gov: NCT05139680.
Keywords: ATTR amyloidosis; ATTR cardiomyopathy; ATTR polyneuropathy; Real-world; Tafamidis.
© 2024. The Author(s).
Conflict of interest statement
Leslie Amass and Rong Wang are employees of Pfizer Inc. and own Pfizer stock. Jennifer M. Stephens, Traci LeMasters, and Rutika Raina are employees of OPEN Health, a contract research organization that received research funding related to this study. Emma Merrill is a clinical research coordinator affiliated with MedStar Health. Nicholas Streicher and Farooq H. Sheikh are investigators affiliated with MedStar Health/Georgetown University School of Medicine, which received funding from Pfizer to support the research.
Figures

References
Associated data
LinkOut - more resources
Full Text Sources
Medical
Research Materials
