

Exploring acetaminophen prodrugs and hybrids: a review
- PMID: 38525062
- PMCID: PMC10958773
- DOI: 10.1039/d4ra00365a
Exploring acetaminophen prodrugs and hybrids: a review
Abstract
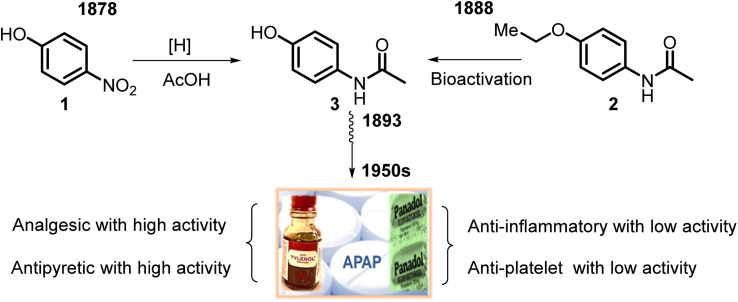
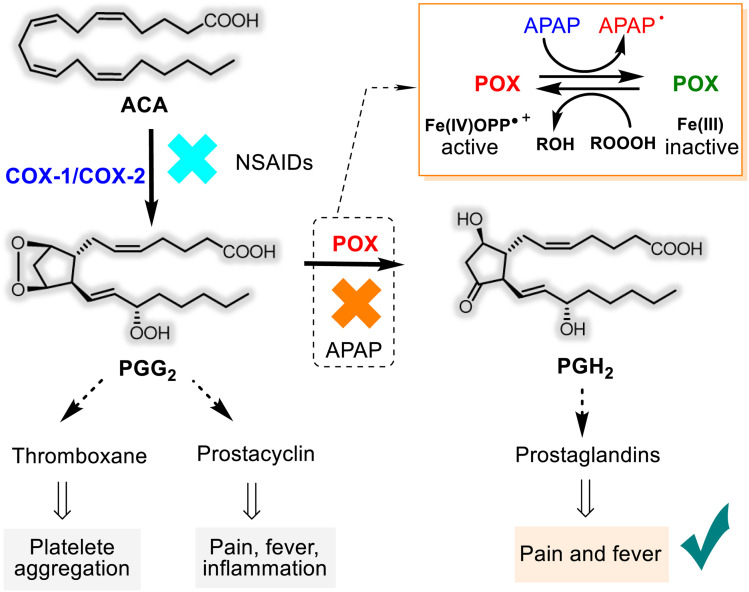
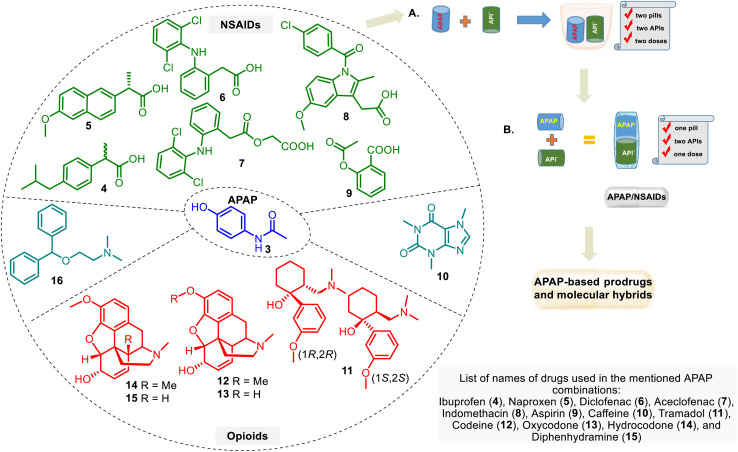
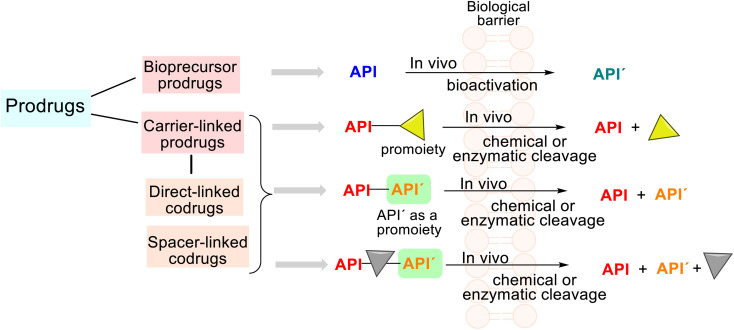
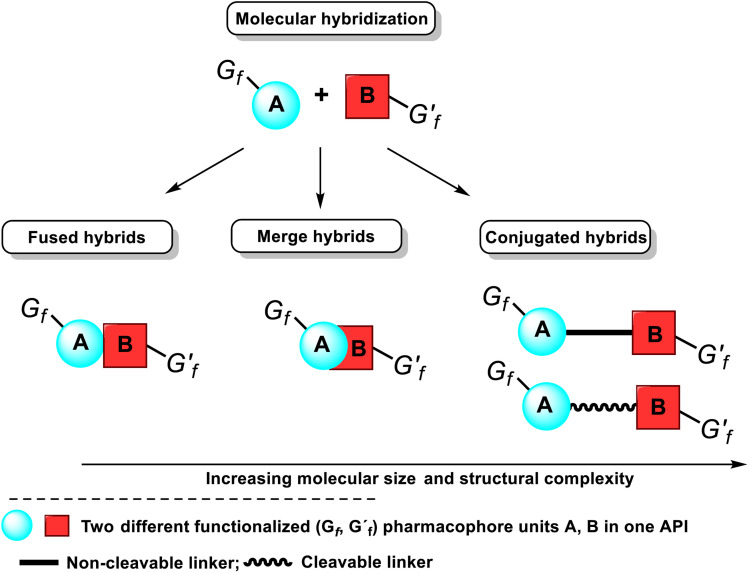
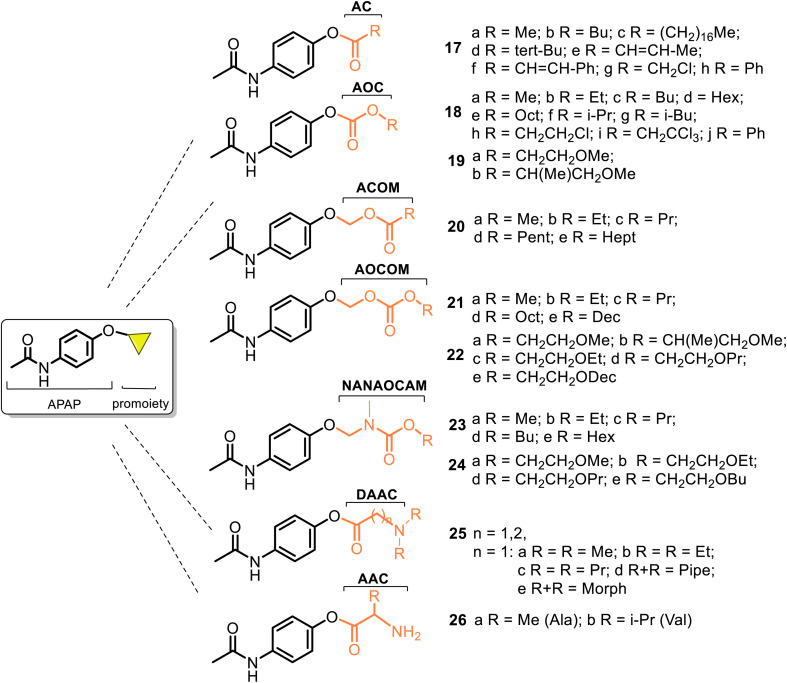
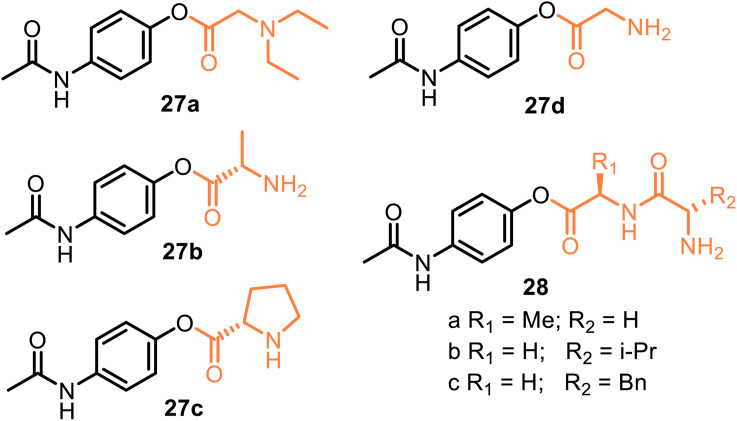
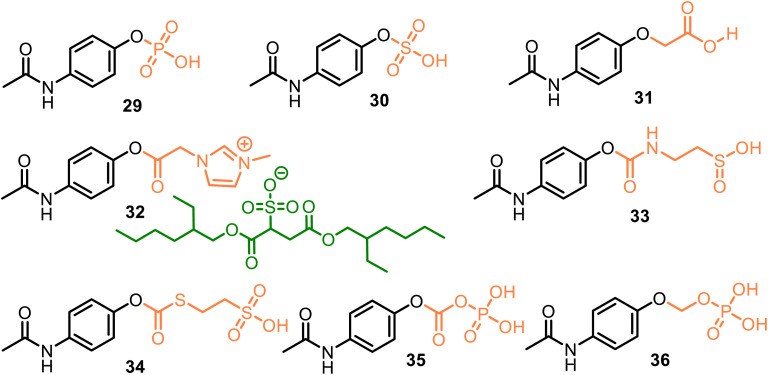
This critical review highlights the advances in developing new molecules for treating pain syndrome, an important issue for human health. Acetaminophen (APAP, known as paracetamol) and nonsteroidal anti-inflammatory drugs (NSAIDs) are commonly used in clinical practice despite their adverse effects. Research is being conducted to develop innovative drugs with improved pharmaceutical properties to mitigate these effects. A more practical way to achieve that is to study well-known and time-tested drugs in their molecular combinations. Accordingly, the present work explores APAP and their combined chemical entities, i.e., prodrugs (soft drugs), codrugs (mutual prodrugs), and hybrids. Due to their molecular structure, APAP prodrugs or codrugs could be considered merged or conjugated hybrids; all these names are very fluid terms. This article proposed a structural classification of these entities to better analyze their advances. So, the following: carrier-linked O-modified APAP, -linked N-modified APAP derivatives (prodrugs), and direct- and spacer-N,O-linked APAP hybrids (codrugs) are the central parts of this review and are examined, especially ester and amide NSAID-APAP molecules. The C-linked APAP and nitric oxide (NO)-releasing APAP hybrids were also briefly discussed. Prime examples of APAP-based drugs such as propacetamol, benorylate, acetaminosalol, nitroparacetamol, and agent JNJ-10450232 weave well into this classification. The proposed classification is the first and original, giving a better understanding of the SAR studies for new pain relievers research and the design development for the analgesic APAP-(or NSAID)-based compounds.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts of interest to declare.
Figures






Similar articles
-
The efficacy of tramadol/acetaminophen combination tablets (Ultracet®) as add-on and maintenance therapy in knee osteoarthritis pain inadequately controlled by nonsteroidal anti-inflammatory drug (NSAID).Clin Rheumatol. 2012 Feb;31(2):317-23. doi: 10.1007/s10067-011-1818-y. Epub 2011 Aug 3. Clin Rheumatol. 2012. PMID: 21811797 Clinical Trial.
-
Propacetamol in dogs: First description of its pharmacokinetics after intravenous and oral administration.Res Vet Sci. 2022 May;144:11-17. doi: 10.1016/j.rvsc.2022.01.002. Epub 2022 Jan 5. Res Vet Sci. 2022. PMID: 35033846
-
Mutual Prodrugs - Codrugs.Curr Med Chem. 2023;30(38):4283-4339. doi: 10.2174/0929867330666221209102650. Curr Med Chem. 2023. PMID: 36503392 Review.
-
Efficacy and Safety of Duloxetine in Patients with Chronic Low Back Pain Who Used versus Did Not Use Concomitant Nonsteroidal Anti-Inflammatory Drugs or Acetaminophen: A Post Hoc Pooled Analysis of 2 Randomized, Placebo-Controlled Trials.Pain Res Treat. 2012;2012:296710. doi: 10.1155/2012/296710. Epub 2012 Mar 18. Pain Res Treat. 2012. PMID: 22550577 Free PMC article.
-
The role of acetaminophen in the treatment of osteoarthritis.Am J Manag Care. 2010 Mar;16 Suppl Management:S48-54. Am J Manag Care. 2010. PMID: 20297877 Review.
References
-
- Blondell R. D. Azadfard M. Wisniewski A. M. Am. Fam. Physician. 2013;87:766. - PubMed
-
- Beal B. R. Wallace M. S. Med. Clin. 2016;100:65. - PubMed
-
- Amaechi O. Human M. M. Featherstone K. Am. Fam. Physician. 2021;104:63. - PubMed
-
- Turk D. C. Wilson H. D. Cahana A. Lancet. 2011;377:2226. - PubMed
-
- Schug S. A. Goddard C. Ann. Palliat. Med. 2014;3:263. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous