

A genome sequence for the threatened whitebark pine
- PMID: 38526344
- PMCID: PMC11075562
- DOI: 10.1093/g3journal/jkae061
A genome sequence for the threatened whitebark pine
Erratum in
-
Correction to: A genome sequence for the threatened whitebark pine.G3 (Bethesda). 2024 Jun 5;14(6):jkae085. doi: 10.1093/g3journal/jkae085. G3 (Bethesda). 2024. PMID: 38683110 Free PMC article. No abstract available.
Abstract
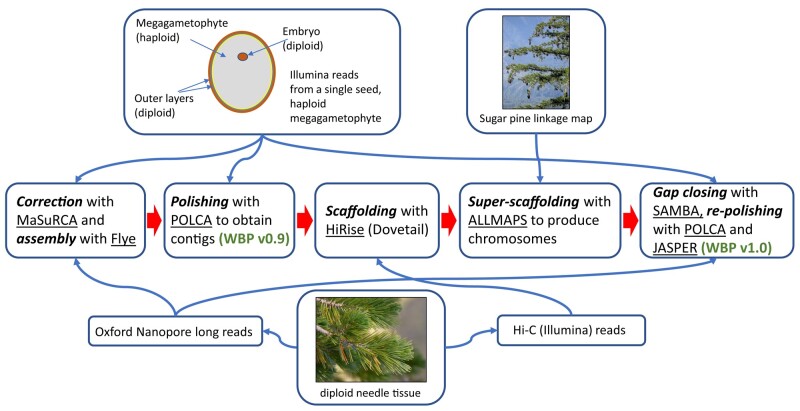
Whitebark pine (WBP, Pinus albicaulis) is a white pine of subalpine regions in the Western contiguous United States and Canada. WBP has become critically threatened throughout a significant part of its natural range due to mortality from the introduced fungal pathogen white pine blister rust (WPBR, Cronartium ribicola) and additional threats from mountain pine beetle (Dendroctonus ponderosae), wildfire, and maladaptation due to changing climate. Vast acreages of WBP have suffered nearly complete mortality. Genomic technologies can contribute to a faster, more cost-effective approach to the traditional practices of identifying disease-resistant, climate-adapted seed sources for restoration. With deep-coverage Illumina short reads of haploid megagametophyte tissue and Oxford Nanopore long reads of diploid needle tissue, followed by a hybrid, multistep assembly approach, we produced a final assembly containing 27.6 Gb of sequence in 92,740 contigs (N50 537,007 bp) and 34,716 scaffolds (N50 2.0 Gb). Approximately 87.2% (24.0 Gb) of total sequence was placed on the 12 WBP chromosomes. Annotation yielded 25,362 protein-coding genes, and over 77% of the genome was characterized as repeats. WBP has demonstrated the greatest variation in resistance to WPBR among the North American white pines. Candidate genes for quantitative resistance include disease resistance genes known as nucleotide-binding leucine-rich repeat receptors (NLRs). A combination of protein domain alignments and direct genome scanning was employed to fully describe the 3 subclasses of NLRs. Our high-quality reference sequence and annotation provide a marked improvement in NLR identification compared to previous assessments that leveraged de novo-assembled transcriptomes.
Keywords: Pinus albicaulis; annotation; conifer; genome assembly; gymnosperm; whitebark pine.
© The Author(s) 2024. Published by Oxford University Press on behalf of The Genetics Society of America.
Conflict of interest statement
Conflicts of interest Any use of product names is for informational purposes only and does not imply endorsement by the US Government. The findings and conclusions in this publication are those of the authors and should not be construed to represent any official USDA or US Government determination or policy.
Figures



Update of
-
A Genome Sequence for the Threatened Whitebark Pine.bioRxiv [Preprint]. 2023 Nov 17:2023.11.16.567420. doi: 10.1101/2023.11.16.567420. bioRxiv. 2023. Update in: G3 (Bethesda). 2024 May 7;14(5):jkae061. doi: 10.1093/g3journal/jkae061. PMID: 38014212 Free PMC article. Updated. Preprint.
References
-
- Bondar EI, Feranchuk SI, Miroshnikova KA, Sharov VV, Kuzmin DA, Oreshkova NV, Krutovsky KV. 2022. Annotation of Siberian larch (Larix sibirica Ledeb.) nuclear genome—one of the most cold-resistant tree species in the only deciduous genus in Pinaceae. Plants (Basel). 11(15):2062. doi: 10.3390/plants11152062. - DOI - PMC - PubMed