

Heterozygous loss-of-function variants in DOCK4 cause neurodevelopmental delay and microcephaly
- PMID: 38526744
- PMCID: PMC11043173
- DOI: 10.1007/s00439-024-02655-4
Heterozygous loss-of-function variants in DOCK4 cause neurodevelopmental delay and microcephaly
Abstract
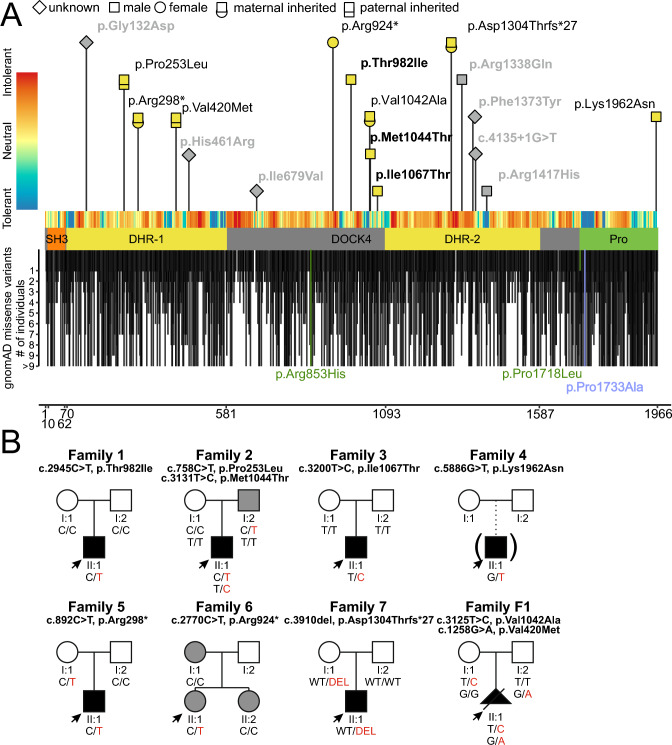
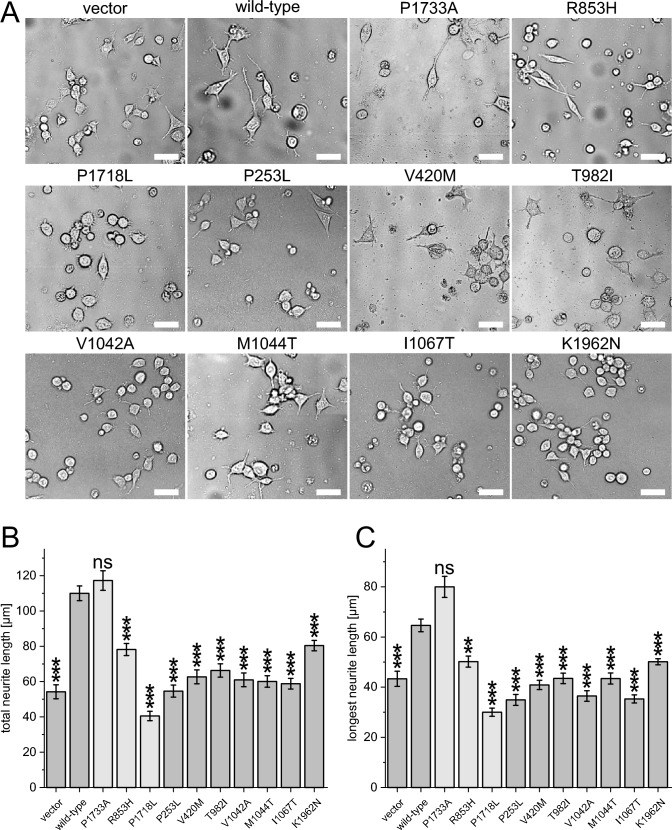
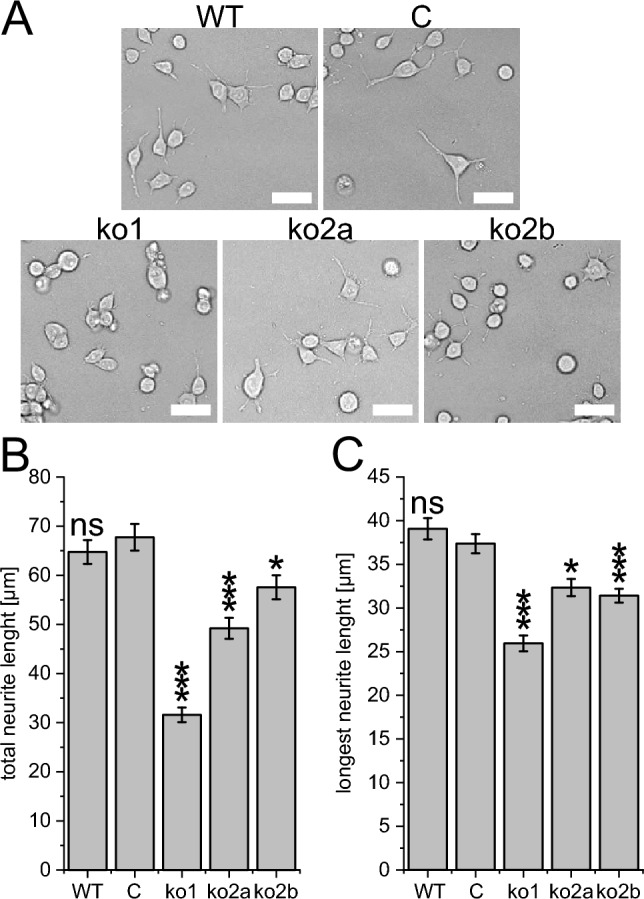
Neurons form the basic anatomical and functional structure of the nervous system, and defects in neuronal differentiation or formation of neurites are associated with various psychiatric and neurodevelopmental disorders. Dynamic changes in the cytoskeleton are essential for this process, which is, inter alia, controlled by the dedicator of cytokinesis 4 (DOCK4) through the activation of RAC1. Here, we clinically describe 7 individuals (6 males and one female) with variants in DOCK4 and overlapping phenotype of mild to severe global developmental delay. Additional symptoms include coordination or gait abnormalities, microcephaly, nonspecific brain malformations, hypotonia and seizures. Four individuals carry missense variants (three of them detected de novo) and three individuals carry null variants (two of them maternally inherited). Molecular modeling of the heterozygous missense variants suggests that the majority of them affect the globular structure of DOCK4. In vitro functional expression studies in transfected Neuro-2A cells showed that all missense variants impaired neurite outgrowth. Furthermore, Dock4 knockout Neuro-2A cells also exhibited defects in promoting neurite outgrowth. Our results, including clinical, molecular and functional data, suggest that loss-of-function variants in DOCK4 probable cause a variable spectrum of a novel neurodevelopmental disorder with microcephaly.
© 2024. The Author(s).
Conflict of interest statement
There are no competing interests.
Figures

References
-
- Abraham S, Scarcia M, Bagshaw RD, McMahon K, Grant G, Harvey T, Yeo M, Esteves FOG, Thygesen HH, Jones PF, Speirs V, Hanby AM, Selby PJ, Lorger M, Dear TN, Pawson T, Marshall CJ, Mavria G. A Rac/Cdc42 exchange factor complex promotes formation of lateral filopodia and blood vessel lumen morphogenesis. Nat Commun. 2015;6:7286. doi: 10.1038/ncomms8286. - DOI - PMC - PubMed
-
- Cacheiro P, Muñoz-Fuentes V, Murray SA, Dickinson ME, Bucan M, Nutter LMJ, Peterson KA, Haselimashhadi H, Flenniken AM, Morgan H, Westerberg H, Konopka T, Hsu C-W, Christiansen A, Lanza DG, Beaudet AL, Heaney JD, Fuchs H, Gailus-Durner V, Sorg T, Prochazka J, Novosadova V, Lelliott CJ, Wardle-Jones H, Wells S, Teboul L, Cater H, Stewart M, Hough T, Wurst W, Sedlacek R, Adams DJ, Seavitt JR, Tocchini-Valentini G, Mammano F, Braun RE, McKerlie C, Herault Y, de Angelis MH, Mallon A-M, Lloyd KCK, Brown SDM, Parkinson H, Meehan TF, Smedley D. Human and mouse essentiality screens as a resource for disease gene discovery. Nat Commun. 2020;11(1):655. doi: 10.1038/s41467-020-14284-2. - DOI - PMC - PubMed
-
- Cioclu MC, Mosca I, Ambrosino P, Puzo D, Bayat A, Wortmann SB, Koch J, Strehlow V, Shirai K, Matsumoto N, Sanders SJ, Michaud V, Legendre M, Riva A, Striano P, Muhle H, Pendziwiat M, Lesca G, Mangano GD, Nardello R, Lemke JR, Møller RS, Soldovieri MV, Rubboli G, Taglialatela M. KCNT2-related disorders: phenotypes, functional, and pharmacological properties. Ann Neurol. 2023;94(2):332–349. doi: 10.1002/ana.26662. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
