

RTN2 deficiency results in an autosomal recessive distal motor neuropathy with lower limb spasticity
- PMID: 38527963
- PMCID: PMC11224604
- DOI: 10.1093/brain/awae091
RTN2 deficiency results in an autosomal recessive distal motor neuropathy with lower limb spasticity
Abstract
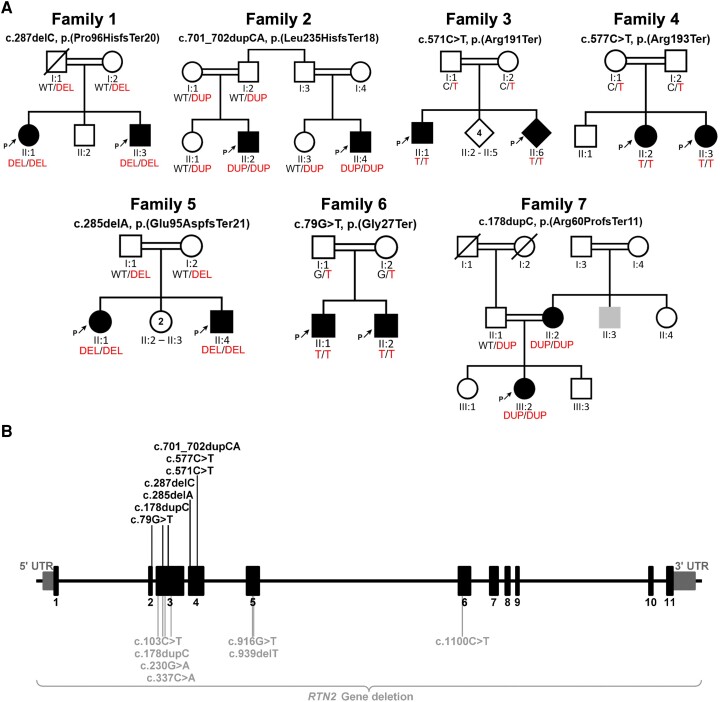
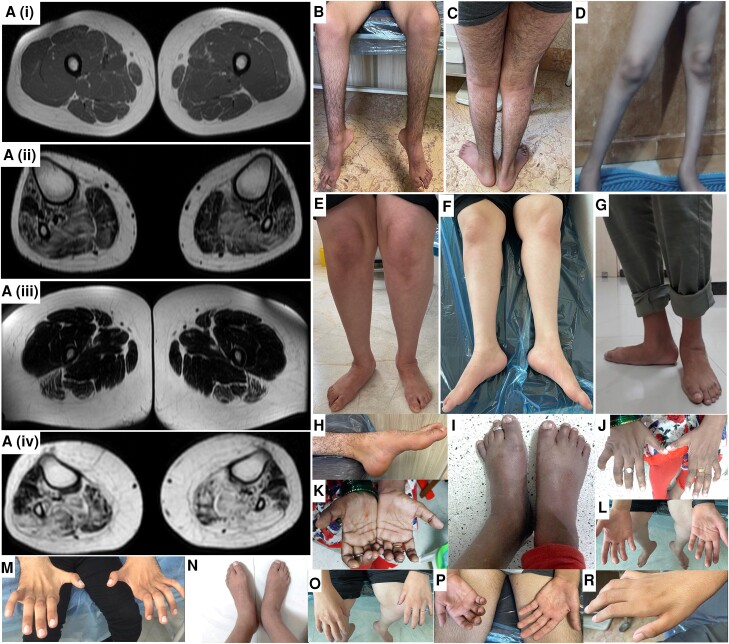
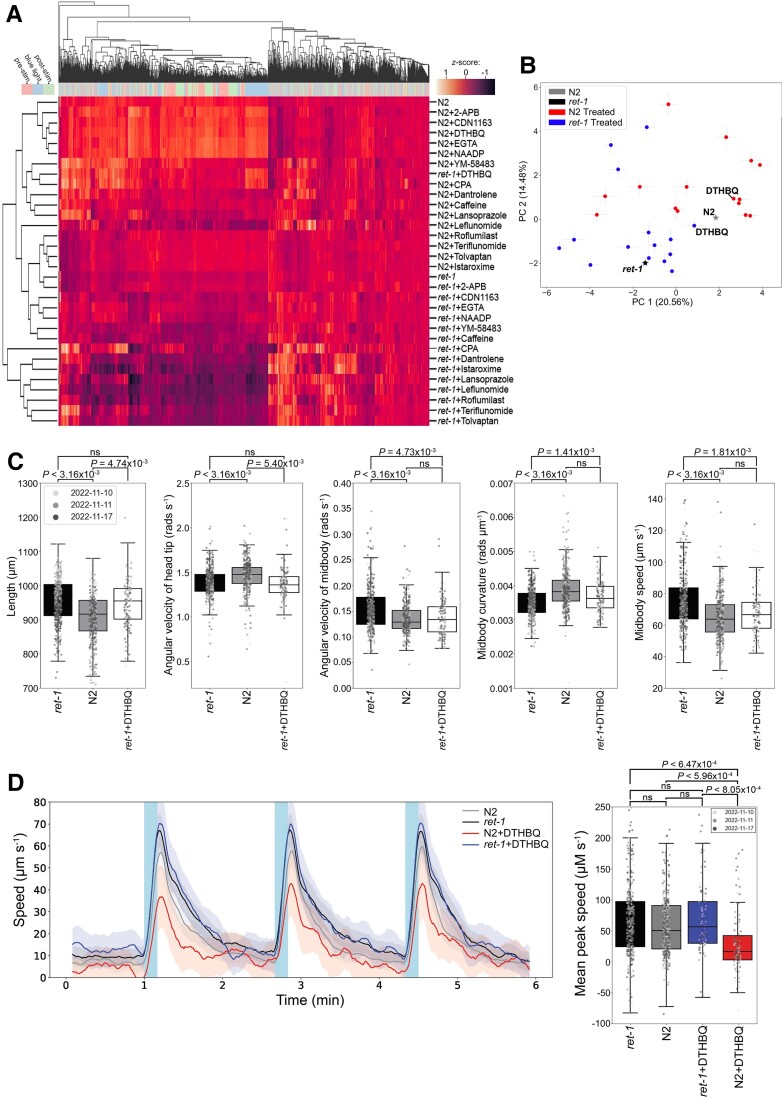
Heterozygous RTN2 variants have been previously identified in a limited cohort of families affected by autosomal dominant spastic paraplegia (SPG12-OMIM:604805) with a variable age of onset. Nevertheless, the definitive validity of SPG12 remains to be confidently confirmed due to the scarcity of supporting evidence. In this study, we identified and validated seven novel or ultra-rare homozygous loss-of-function RTN2 variants in 14 individuals from seven consanguineous families with distal hereditary motor neuropathy (dHMN) using exome, genome and Sanger sequencing coupled with deep-phenotyping. All affected individuals (seven males and seven females, aged 9-50 years) exhibited weakness in the distal upper and lower limbs, lower limb spasticity and hyperreflexia, with onset in the first decade of life. Nerve conduction studies revealed axonal motor neuropathy with neurogenic changes in the electromyography. Despite a slowly progressive disease course, all patients remained ambulatory over a mean disease duration of 19.71 ± 13.70 years. Characterization of Caenorhabditis elegans RTN2 homologous loss-of-function variants demonstrated morphological and behavioural differences compared with the parental strain. Treatment of the mutant with an endoplasmic/sarcoplasmic reticulum Ca2+ reuptake inhibitor (2,5-di-tert-butylhydroquinone) rescued key phenotypic differences, suggesting a potential therapeutic benefit for RTN2-disorder. Despite RTN2 being an endoplasmic reticulum (ER)-resident membrane shaping protein, our analysis of patient fibroblast cells did not find significant alterations in ER structure or the response to ER stress. Our findings delineate a distinct form of autosomal recessive dHMN with pyramidal features associated with RTN2 deficiency. This phenotype shares similarities with SIGMAR1-related dHMN and Silver-like syndromes, providing valuable insights into the clinical spectrum and potential therapeutic strategies for RTN2-related dHMN.
Keywords: dHMN; hereditary spastic paraplegia; neurodegeneration; polyneuropathy.
© The Author(s) 2024. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
G.Z. and P.B. are employees of CENTOGENE. The other authors report no competing interests.
Figures
References
MeSH terms
Grants and funding
- Michael J Fox Foundation for Parkinson's Research
- Charcot-Marie-Tooth Association
- Multiple System Atrophy Trust
- National Institute for Health Research
- U54 NS065712/NS/NINDS NIH HHS/United States
- HU 800/15-1/Deutsche Forschungsgemeinschaft
- MC-A658-5TY30/WT_/Wellcome Trust/United Kingdom
- MDA510281/Muscular Dystrophy Association
- Sparks
- Brain Research UK
- Great Ormond Street Hospital Charity
- ERC_/European Research Council/International
- U54NS065712/NS/NINDS NIH HHS/United States
- BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 714853/Horizon 2020
- UCLH Biomedical Research Centre
- Ataxia UK
- WT_/Wellcome Trust/United Kingdom
- MR/S005021/1/MRC_/Medical Research Council/United Kingdom
- Alzheimer's Research UK
- DAAD
- Rosetrees Trust
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
