

Fast and flexible profiling of chromatin accessibility and total RNA expression in single nuclei using Microwell-seq3
- PMID: 38531851
- PMCID: PMC10966074
- DOI: 10.1038/s41421-023-00642-z
Fast and flexible profiling of chromatin accessibility and total RNA expression in single nuclei using Microwell-seq3
Abstract
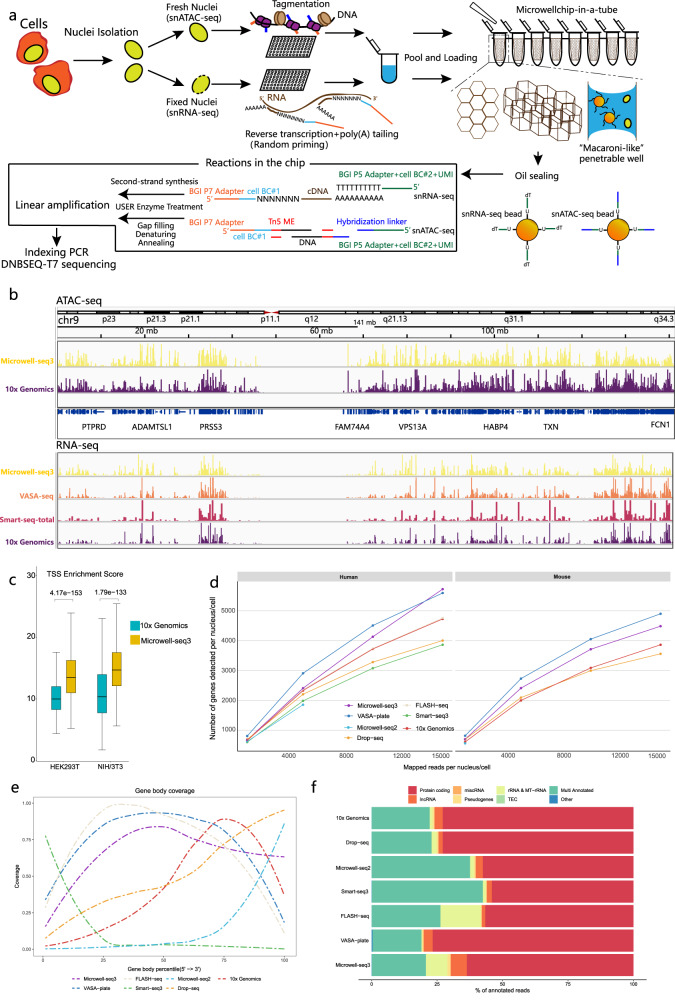
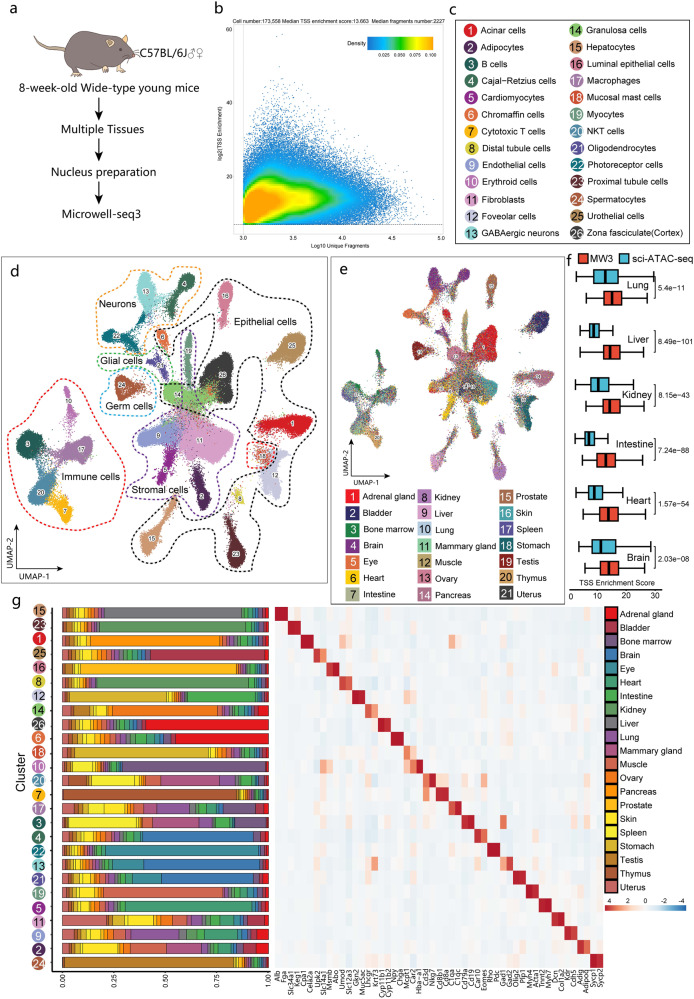
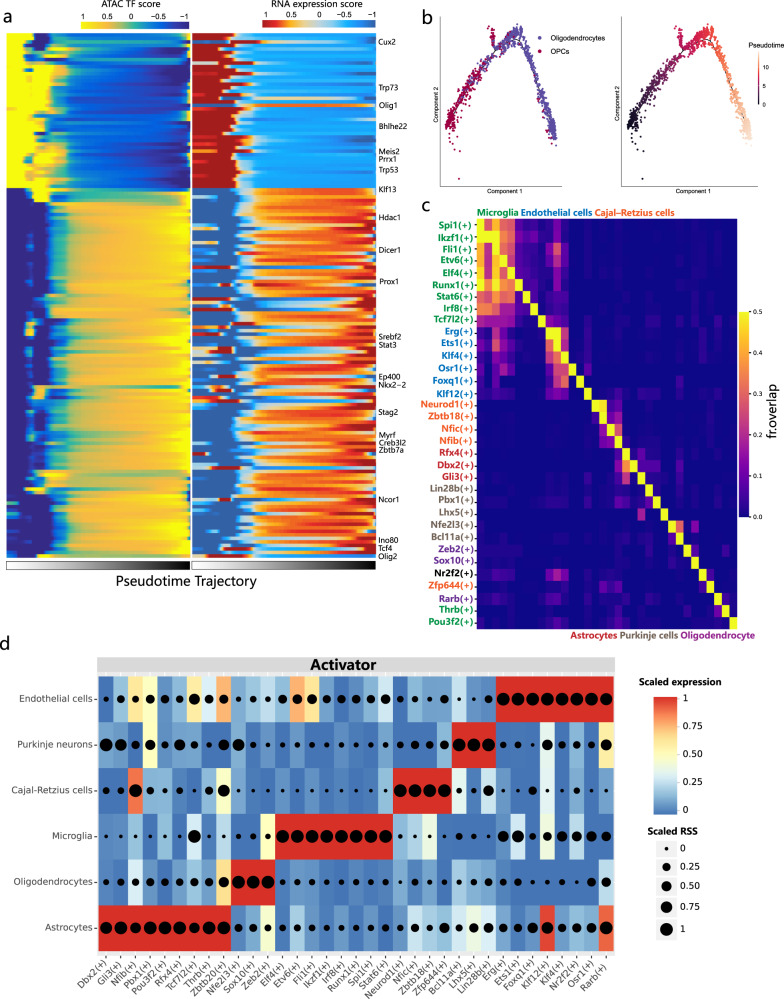
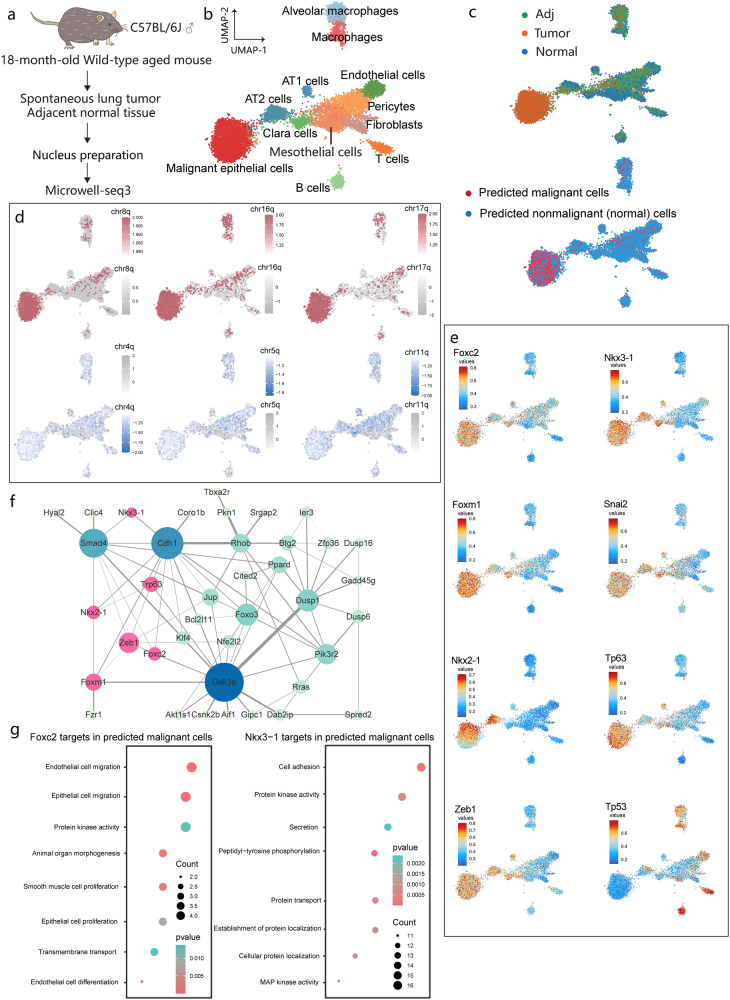
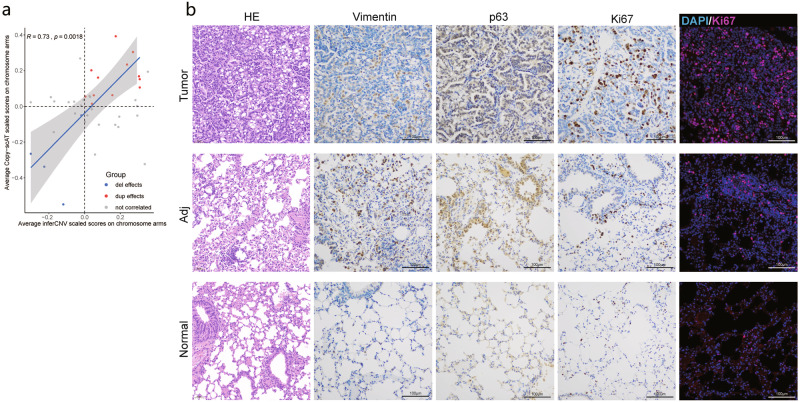
Single cell chromatin accessibility profiling and transcriptome sequencing are the most widely used technologies for single-cell genomics. Here, we present Microwell-seq3, a high-throughput and facile platform for high-sensitivity single-nucleus chromatin accessibility or full-length transcriptome profiling. The method combines a preindexing strategy and a penetrable chip-in-a-tube for single nucleus loading and DNA amplification and therefore does not require specialized equipment. We used Microwell-seq3 to profile chromatin accessibility in more than 200,000 single nuclei and the full-length transcriptome in ~50,000 nuclei from multiple adult mouse tissues. Compared with the existing polyadenylated transcript capture methods, integrative analysis of cell type-specific regulatory elements and total RNA expression uncovered comprehensive cell type heterogeneity in the brain. Gene regulatory networks based on chromatin accessibility profiling provided an improved cell type communication model. Finally, we demonstrated that Microwell-seq3 can identify malignant cells and their specific regulons in spontaneous lung tumors of aged mice. We envision a broad application of Microwell-seq3 in many areas of research.
© 2024. The Author(s).
Conflict of interest statement
G.G., F.Y., G.Z., L.Y. and X.H. have filed a patent application related to this work. The remaining authors declare no competing interests.
Figures
Similar articles
-
Single-cell sequencing of full-length transcripts and T-cell receptors with automated high-throughput Smart-seq3.BMC Genomics. 2024 Nov 21;25(1):1127. doi: 10.1186/s12864-024-11036-0. BMC Genomics. 2024. PMID: 39574011 Free PMC article.
-
Hydrop enables droplet-based single-cell ATAC-seq and single-cell RNA-seq using dissolvable hydrogel beads.Elife. 2022 Feb 23;11:e73971. doi: 10.7554/eLife.73971. Elife. 2022. PMID: 35195064 Free PMC article.
-
ISSAAC-seq enables sensitive and flexible multimodal profiling of chromatin accessibility and gene expression in single cells.Nat Methods. 2022 Oct;19(10):1243-1249. doi: 10.1038/s41592-022-01601-4. Epub 2022 Sep 15. Nat Methods. 2022. PMID: 36109677
-
A plate-based single-cell ATAC-seq workflow for fast and robust profiling of chromatin accessibility.Nat Protoc. 2021 Aug;16(8):4084-4107. doi: 10.1038/s41596-021-00583-5. Epub 2021 Jul 19. Nat Protoc. 2021. PMID: 34282334 Review.
-
Application of Single-Cell Assay for Transposase-Accessible Chromatin with High Throughput Sequencing in Plant Science: Advances, Technical Challenges, and Prospects.Int J Mol Sci. 2024 Jan 25;25(3):1479. doi: 10.3390/ijms25031479. Int J Mol Sci. 2024. PMID: 38338756 Free PMC article. Review.
Cited by
-
Advances and applications in single-cell and spatial genomics.Sci China Life Sci. 2025 May;68(5):1226-1282. doi: 10.1007/s11427-024-2770-x. Epub 2024 Dec 20. Sci China Life Sci. 2025. PMID: 39792333 Review.
-
Time-resolved single-cell transcriptomic sequencing.Chem Sci. 2024 Oct 30;15(46):19225-19246. doi: 10.1039/d4sc05700g. eCollection 2024 Nov 27. Chem Sci. 2024. PMID: 39568874 Free PMC article. Review.
-
Automated high-throughput profiling of single-cell total transcriptome with scComplete-seq.Nucleic Acids Res. 2025 Jul 19;53(14):gkaf699. doi: 10.1093/nar/gkaf699. Nucleic Acids Res. 2025. PMID: 40682827 Free PMC article.
-
Chromatin accessibility: biological functions, molecular mechanisms and therapeutic application.Signal Transduct Target Ther. 2024 Dec 4;9(1):340. doi: 10.1038/s41392-024-02030-9. Signal Transduct Target Ther. 2024. PMID: 39627201 Free PMC article. Review.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
