

Expanding the clinical spectrum of biglycan-related Meester-Loeys syndrome
- PMID: 38531898
- PMCID: PMC10966070
- DOI: 10.1038/s41525-024-00413-z
Expanding the clinical spectrum of biglycan-related Meester-Loeys syndrome
Abstract
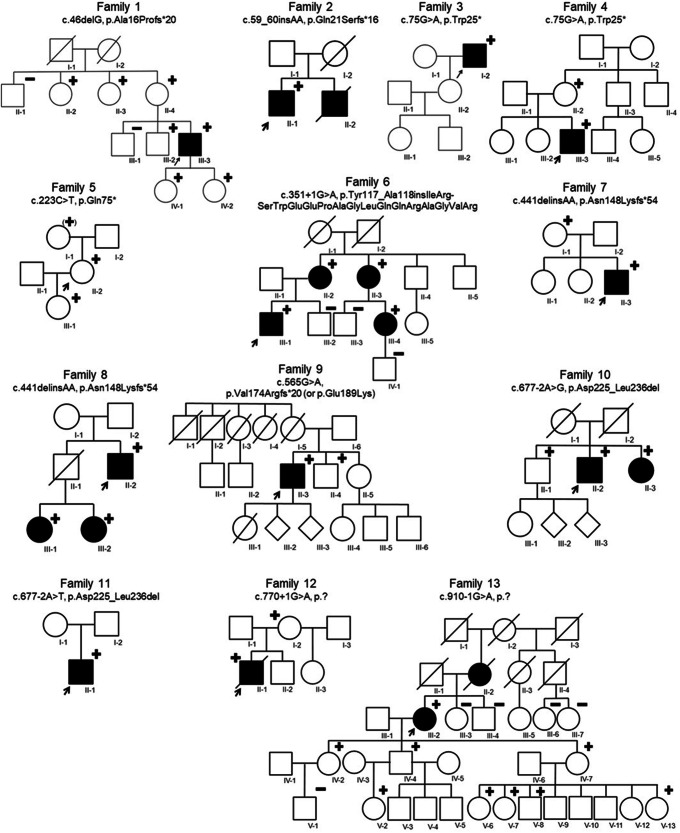
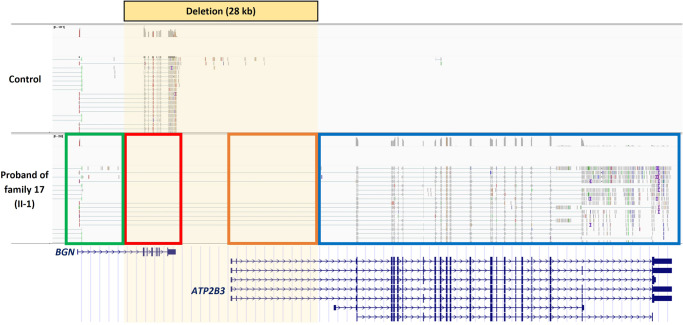
Pathogenic loss-of-function variants in BGN, an X-linked gene encoding biglycan, are associated with Meester-Loeys syndrome (MRLS), a thoracic aortic aneurysm/dissection syndrome. Since the initial publication of five probands in 2017, we have considerably expanded our MRLS cohort to a total of 18 probands (16 males and 2 females). Segregation analyses identified 36 additional BGN variant-harboring family members (9 males and 27 females). The identified BGN variants were shown to lead to loss-of-function by cDNA and Western Blot analyses of skin fibroblasts or were strongly predicted to lead to loss-of-function based on the nature of the variant. No (likely) pathogenic missense variants without additional (predicted) splice effects were identified. Interestingly, a male proband with a deletion spanning the coding sequence of BGN and the 5' untranslated region of the downstream gene (ATP2B3) presented with a more severe skeletal phenotype. This may possibly be explained by expressional activation of the downstream ATPase ATP2B3 (normally repressed in skin fibroblasts) driven by the remnant BGN promotor. This study highlights that aneurysms and dissections in MRLS extend beyond the thoracic aorta, affecting the entire arterial tree, and cardiovascular symptoms may coincide with non-specific connective tissue features. Furthermore, the clinical presentation is more severe and penetrant in males compared to females. Extensive analysis at RNA, cDNA, and/or protein level is recommended to prove a loss-of-function effect before determining the pathogenicity of identified BGN missense and non-canonical splice variants. In conclusion, distinct mechanisms may underlie the wide phenotypic spectrum of MRLS patients carrying loss-of-function variants in BGN.
© 2024. The Author(s).
Conflict of interest statement
S.A.B. is an employee of GeneDx, LLC. All other authors declare no conflict of interest.
Figures

References
-
- Marfan A-B. Un cas de deformation congenitales des quatre membres plus prononcee aux extremities characterisee par l’allongment des os avec un certain dgre d’amincissement. Bull. Mem. Soc. Med. Hop. (Paris) 1986;13:220.
-
- HC D. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Am. J. Hum. Genet. 1991;49:662–667. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
