

Paradoxical Activation of Oncogenic Signaling as a Cancer Treatment Strategy
- PMID: 38533987
- PMCID: PMC11215412
- DOI: 10.1158/2159-8290.CD-23-0216
Paradoxical Activation of Oncogenic Signaling as a Cancer Treatment Strategy
Abstract
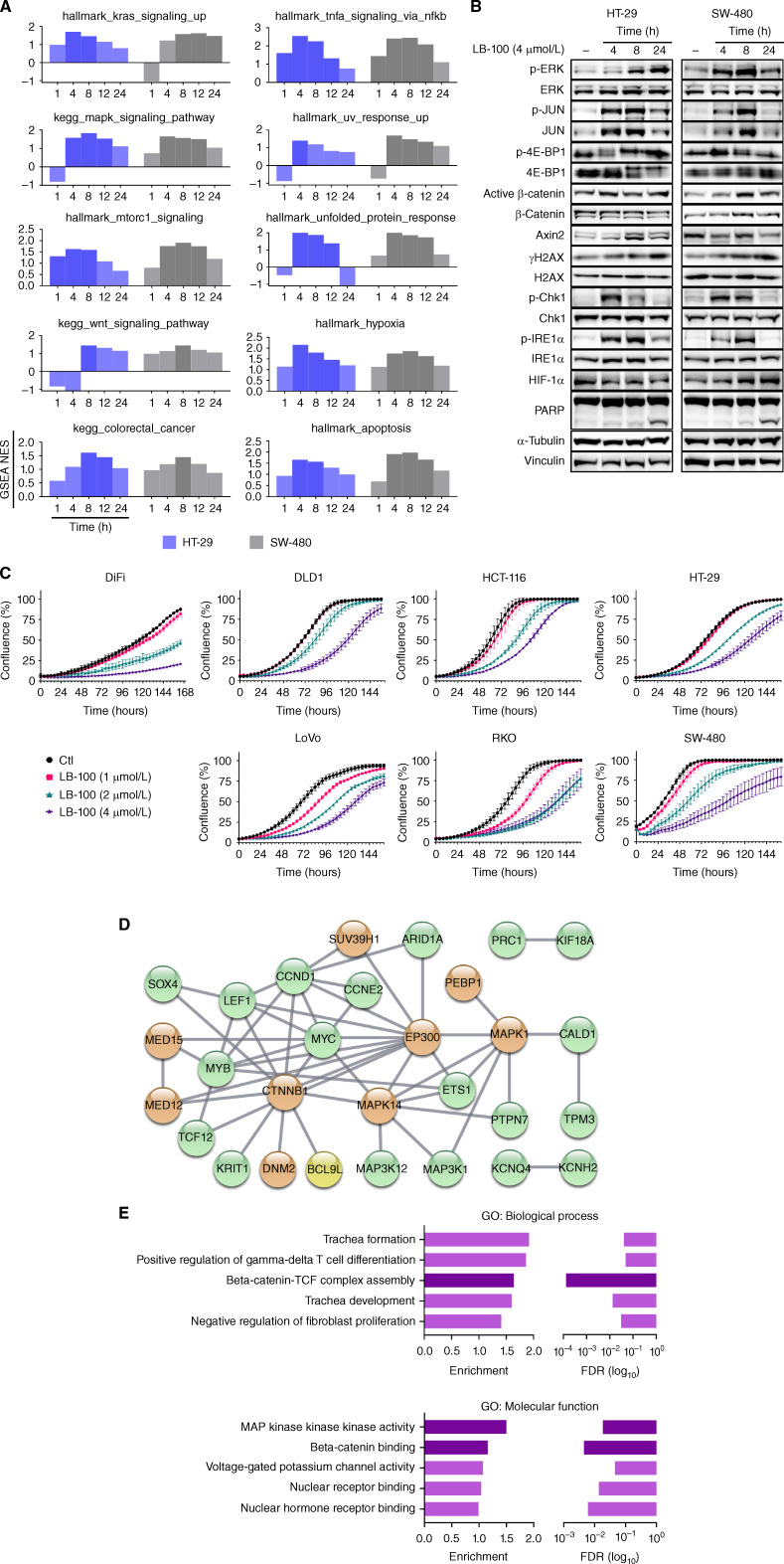
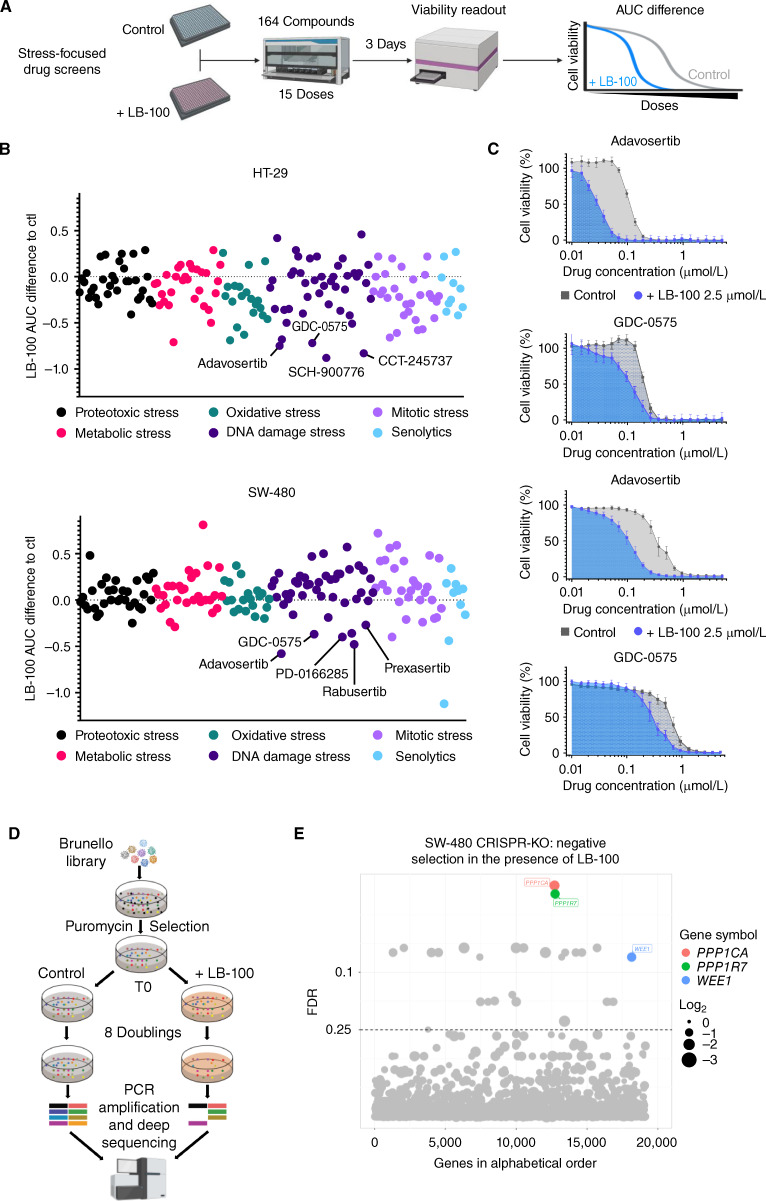
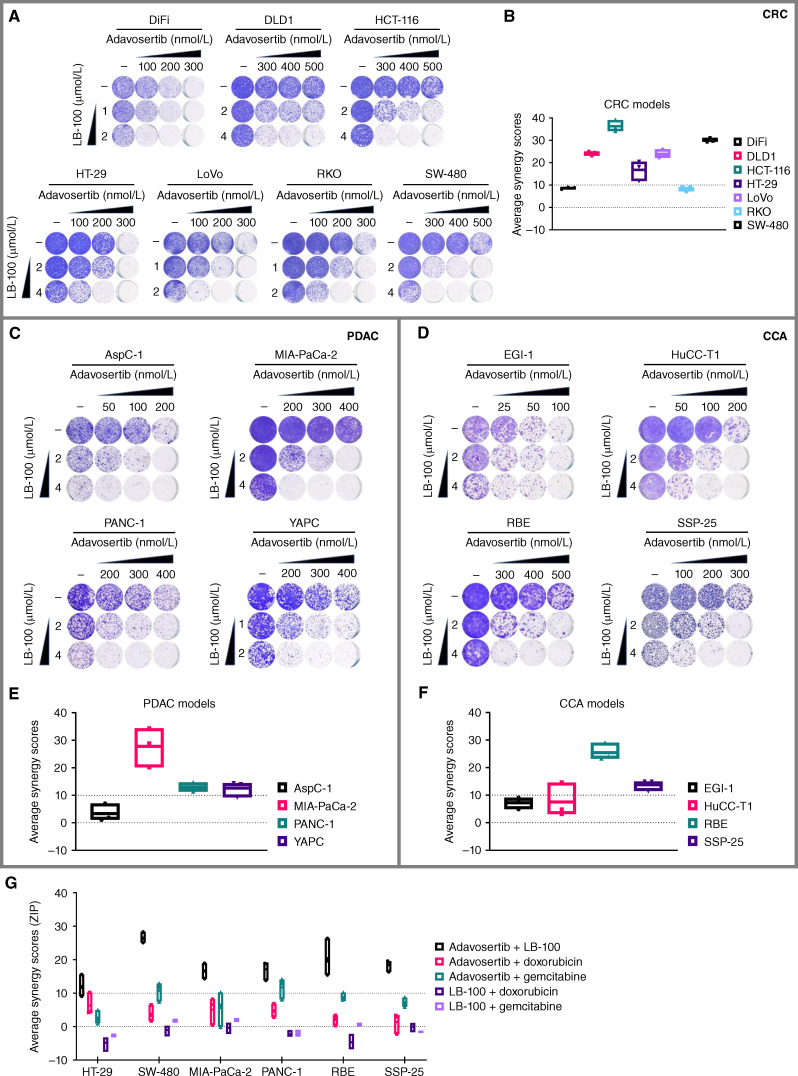
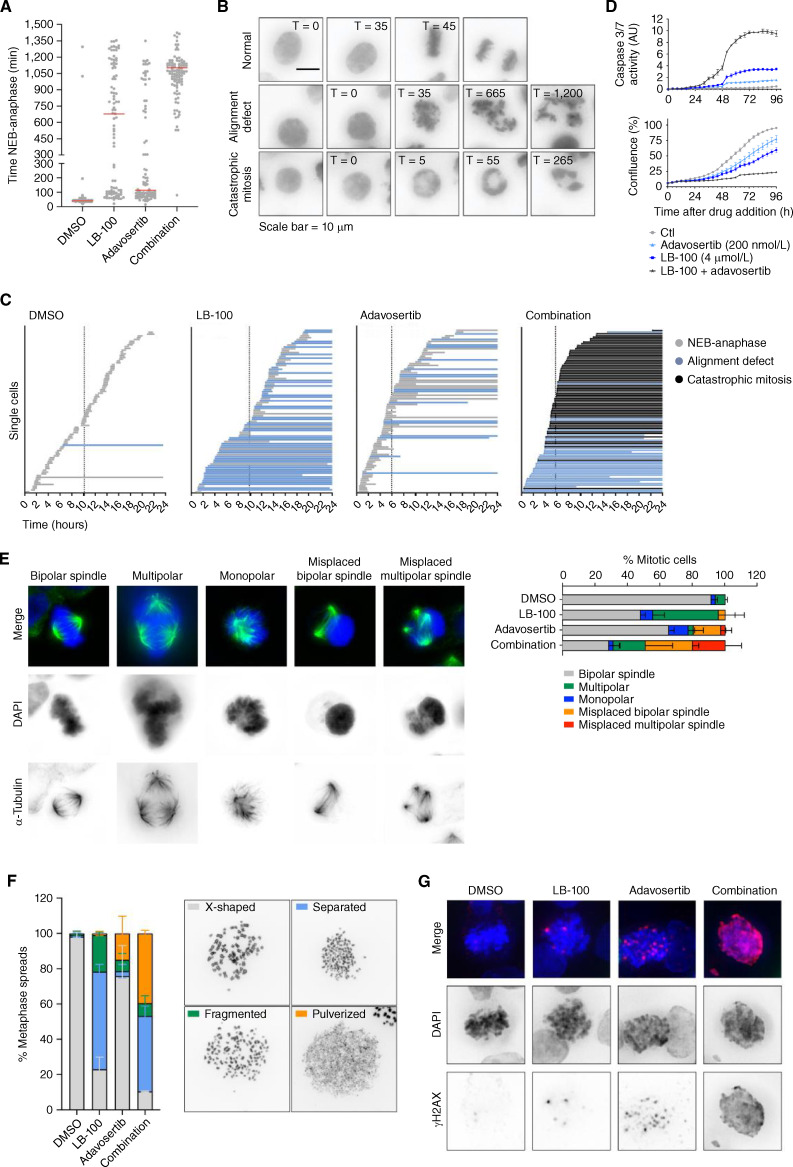
Cancer homeostasis depends on a balance between activated oncogenic pathways driving tumorigenesis and engagement of stress response programs that counteract the inherent toxicity of such aberrant signaling. Although inhibition of oncogenic signaling pathways has been explored extensively, there is increasing evidence that overactivation of the same pathways can also disrupt cancer homeostasis and cause lethality. We show here that inhibition of protein phosphatase 2A (PP2A) hyperactivates multiple oncogenic pathways and engages stress responses in colon cancer cells. Genetic and compound screens identify combined inhibition of PP2A and WEE1 as synergistic in multiple cancer models by collapsing DNA replication and triggering premature mitosis followed by cell death. This combination also suppressed the growth of patient-derived tumors in vivo. Remarkably, acquired resistance to this drug combination suppressed the ability of colon cancer cells to form tumors in vivo. Our data suggest that paradoxical activation of oncogenic signaling can result in tumor-suppressive resistance. Significance: A therapy consisting of deliberate hyperactivation of oncogenic signaling combined with perturbation of the stress responses that result from this is very effective in animal models of colon cancer. Resistance to this therapy is associated with loss of oncogenic signaling and reduced oncogenic capacity, indicative of tumor-suppressive drug resistance.
©2024 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
M.H. Dias has a patent for drug combination pending. R. Bernards reports grants from Lixte Biotechnology during the conduct of the study; personal fees from Lixte Biotechnology outside the submitted work; in addition, R. Bernards has a patent for Application pending. No disclosures were reported by the other authors.
Figures
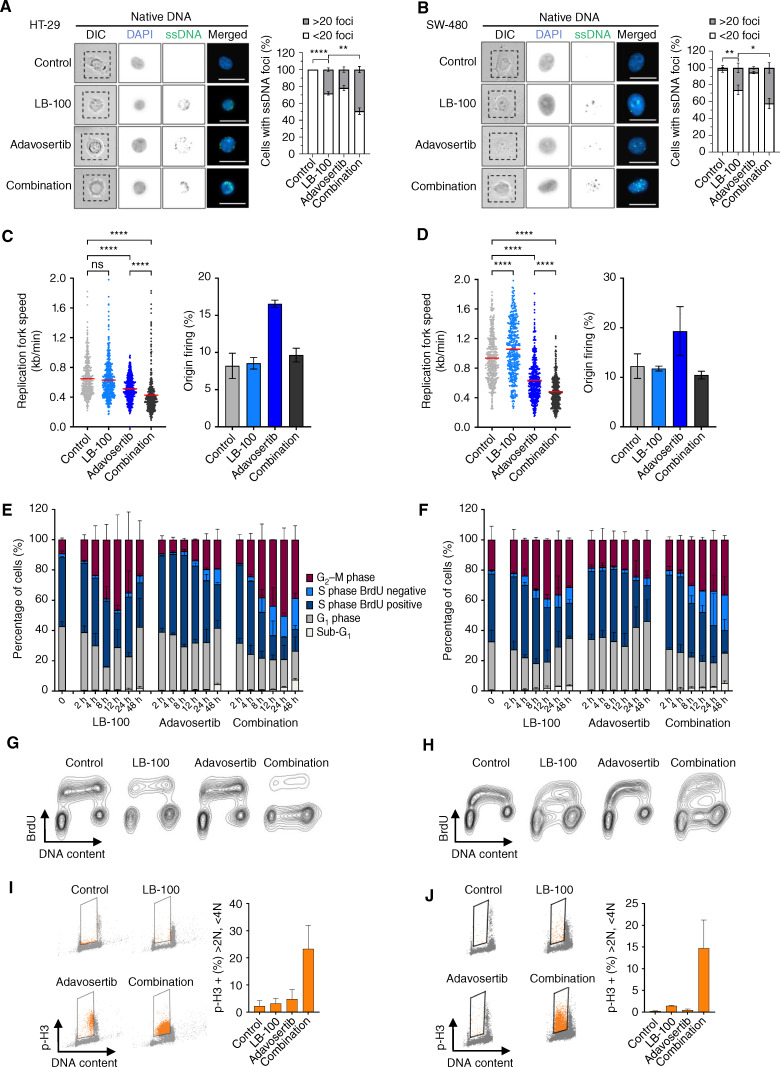
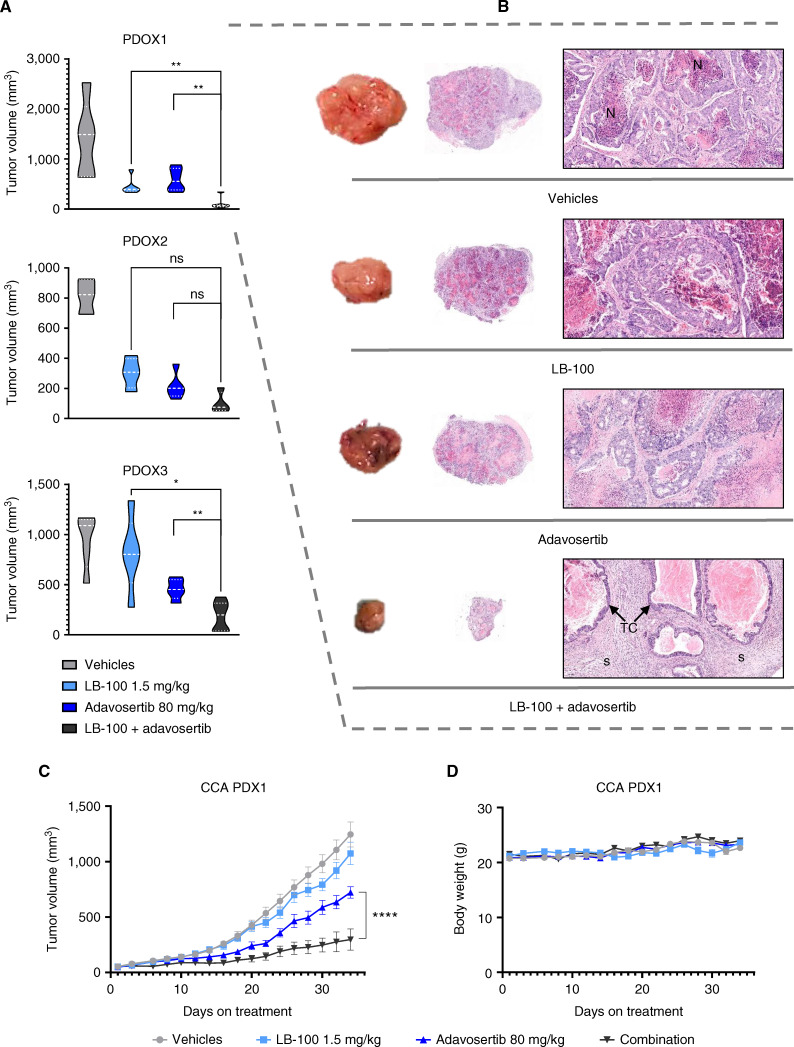
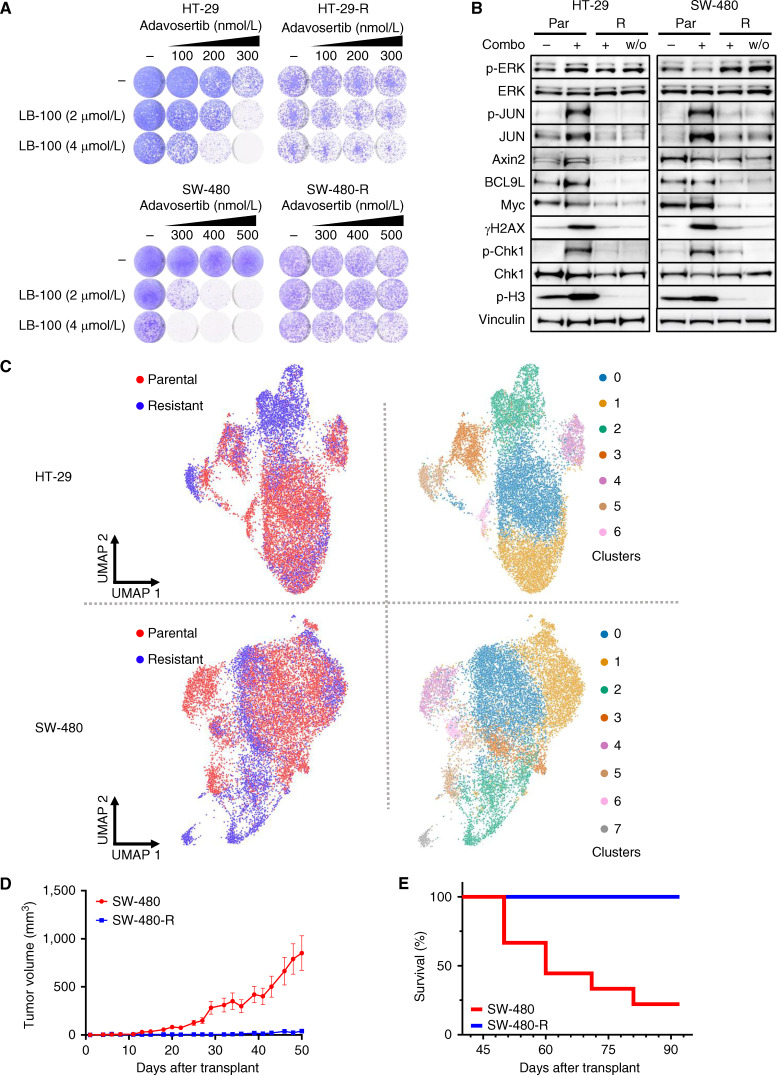
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
