

The GTPase activating protein Gyp7 regulates Rab7/Ypt7 activity on late endosomes
- PMID: 38536036
- PMCID: PMC10978497
- DOI: 10.1083/jcb.202305038
The GTPase activating protein Gyp7 regulates Rab7/Ypt7 activity on late endosomes
Abstract
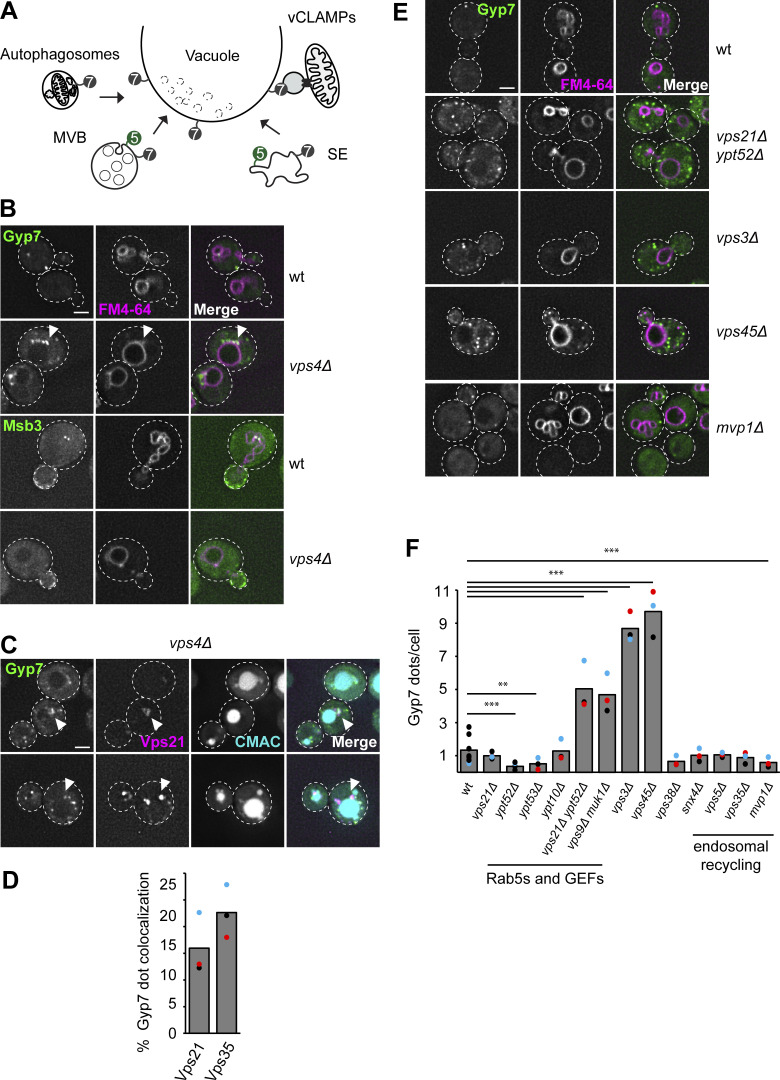
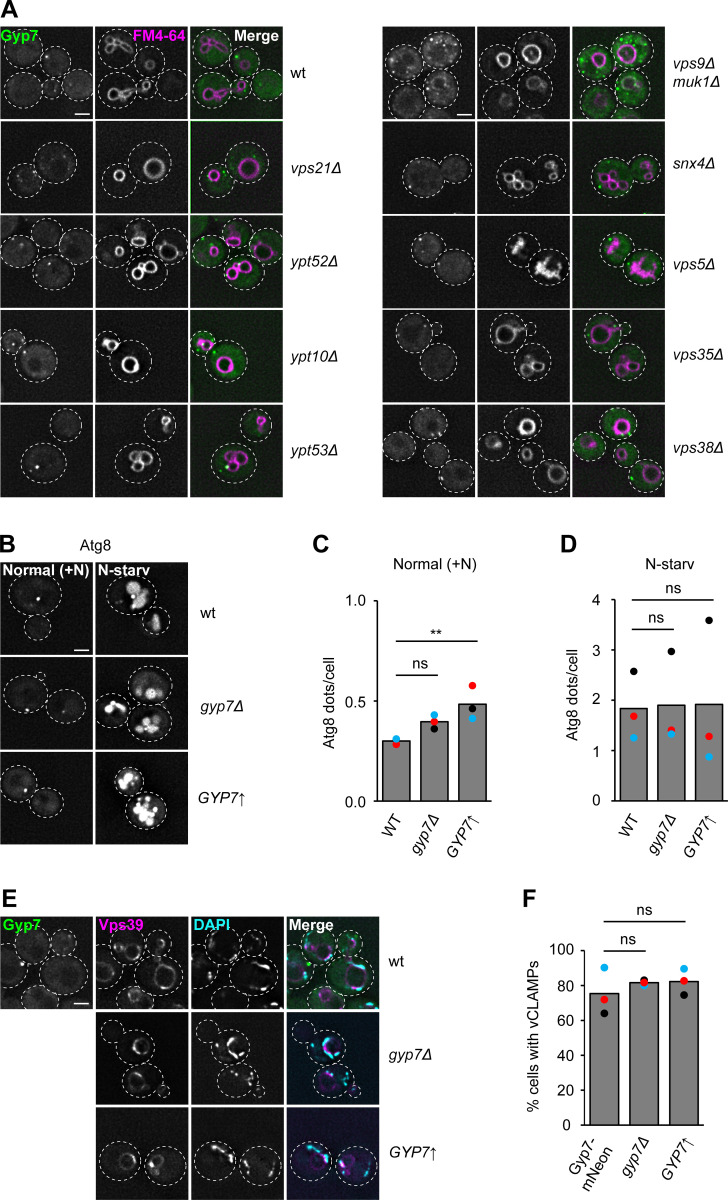
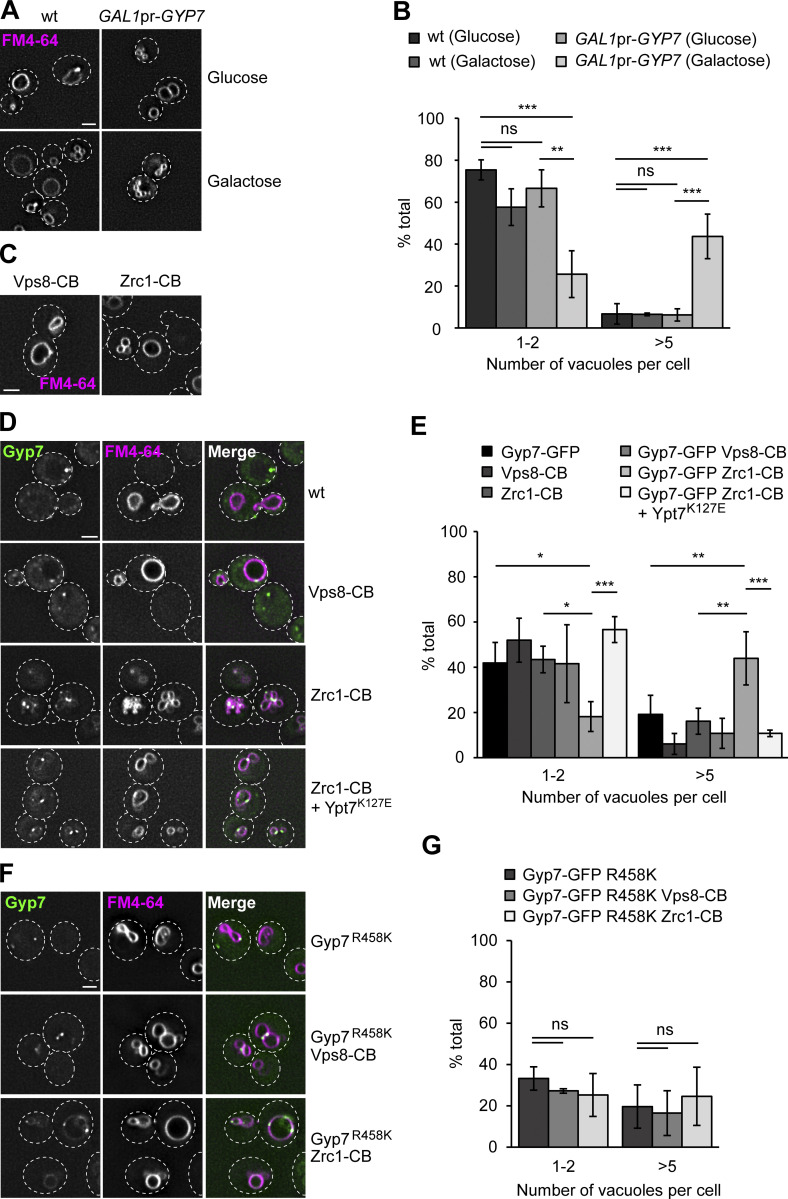
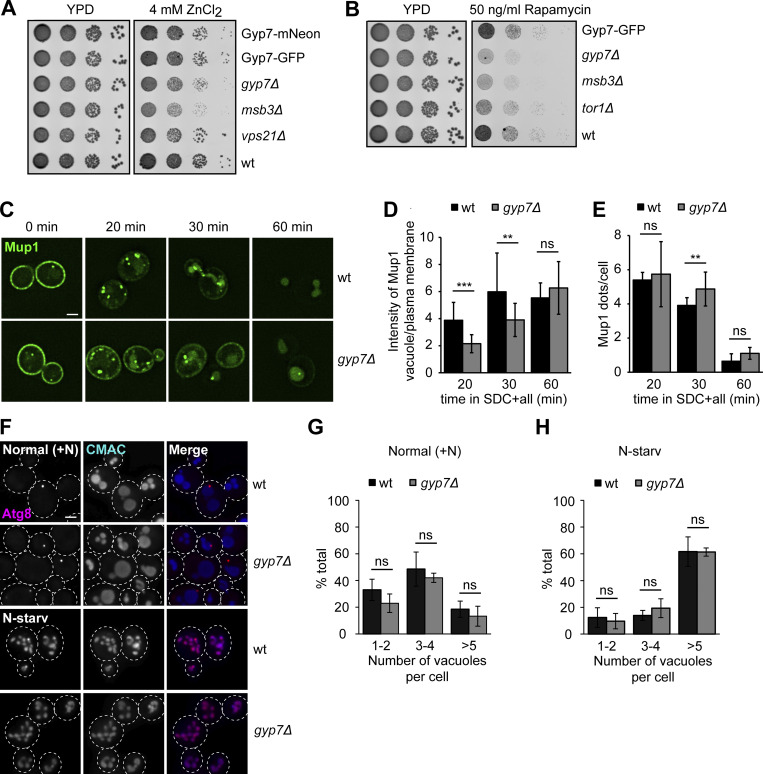
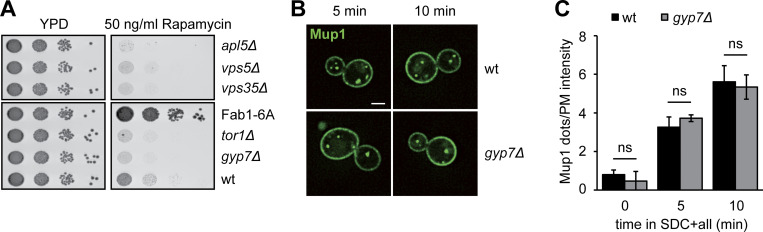
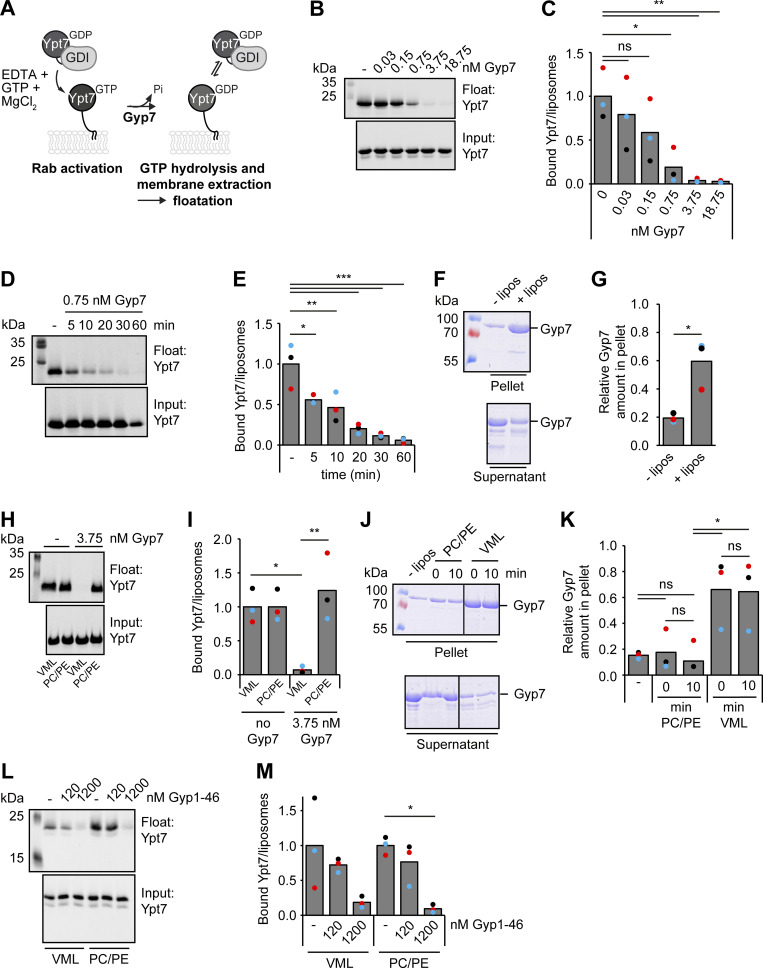
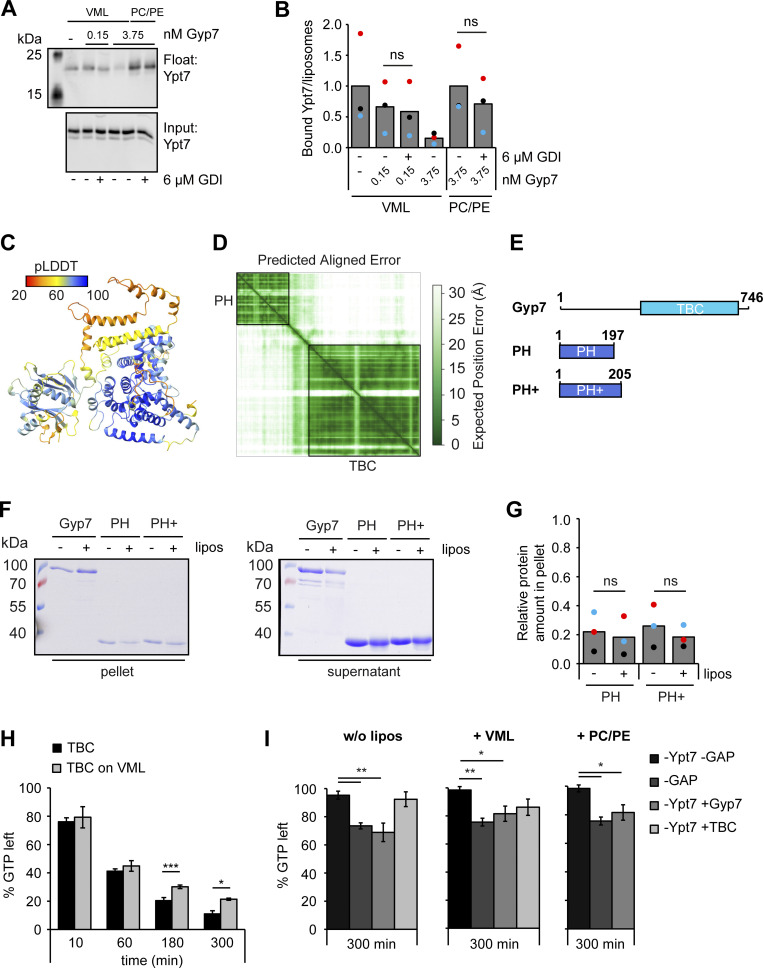
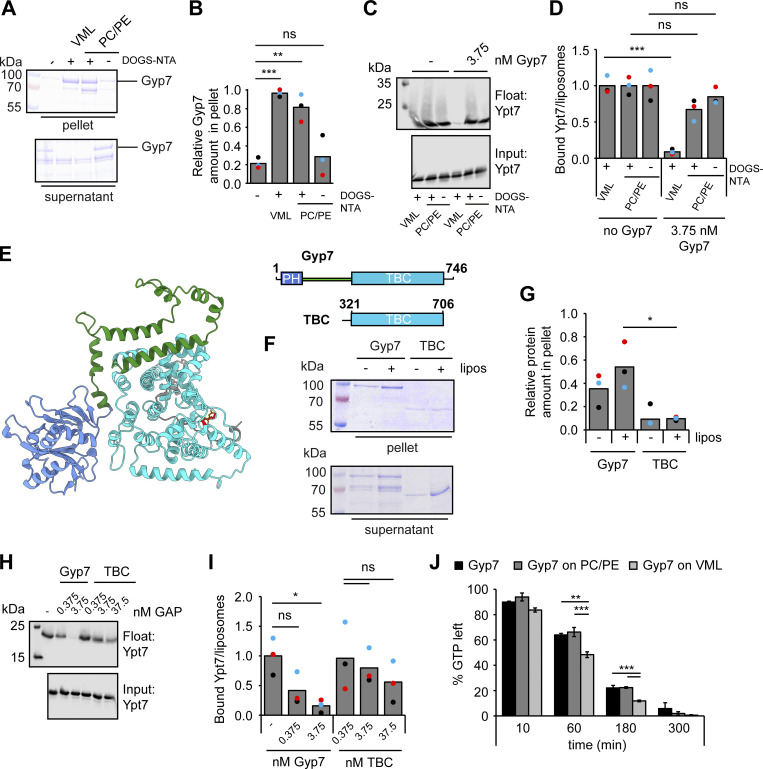
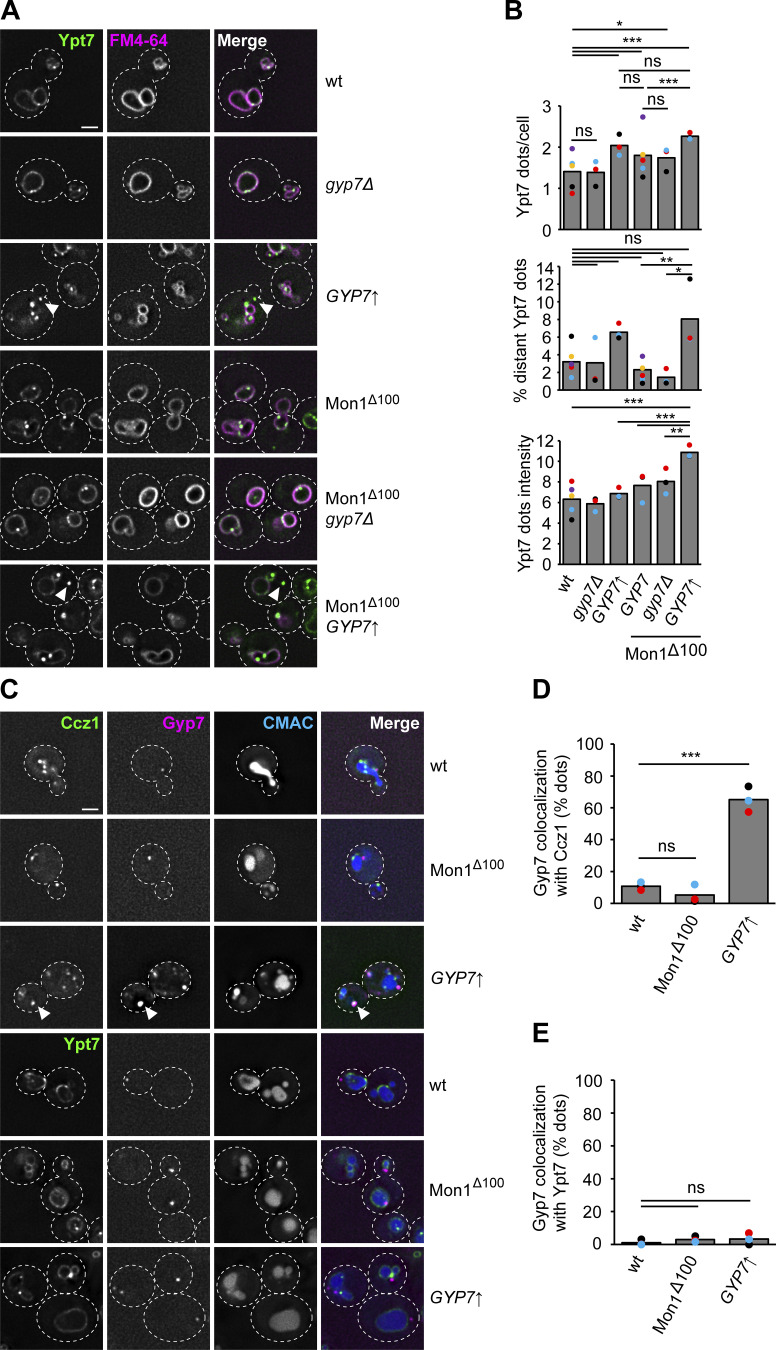
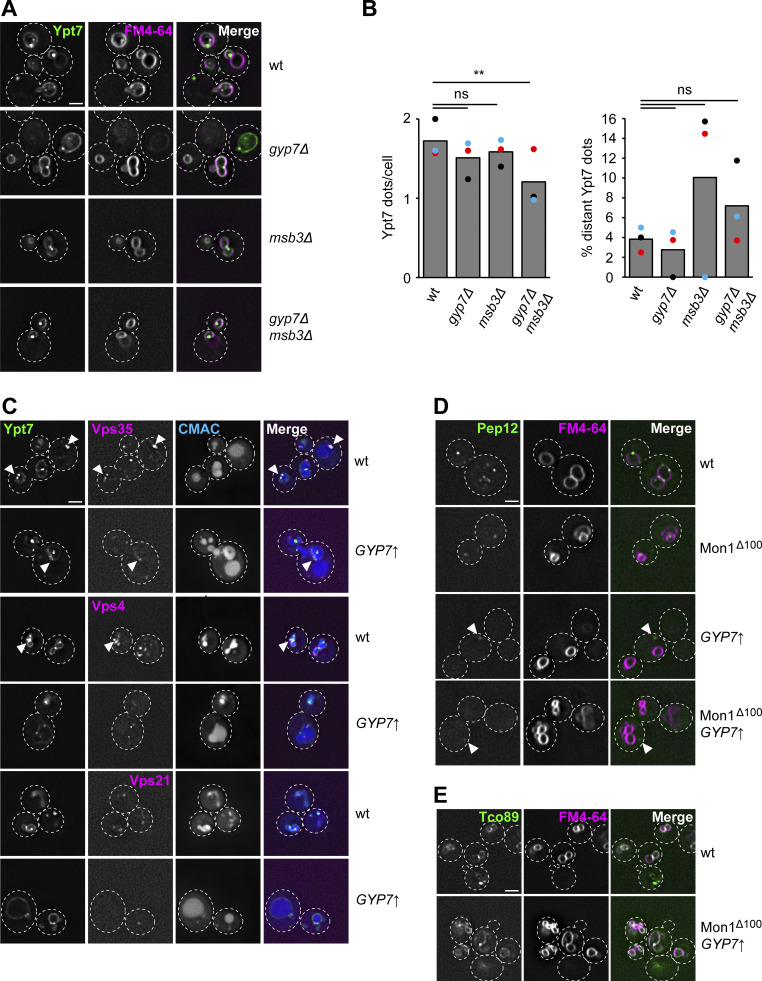
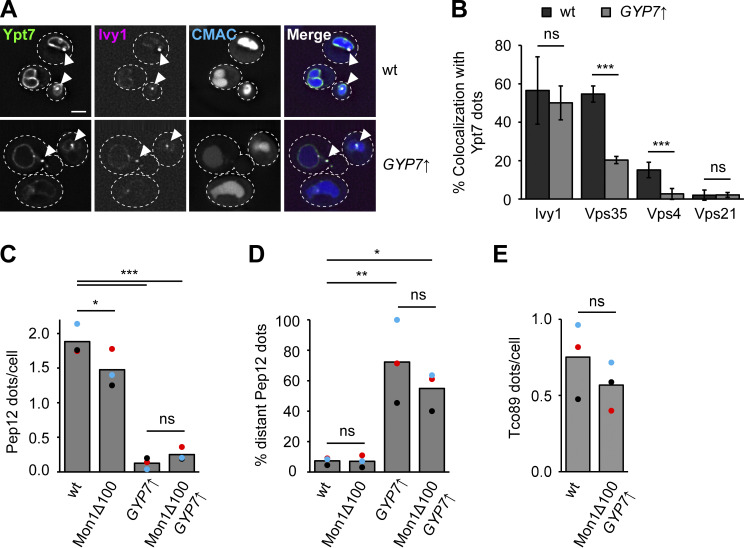
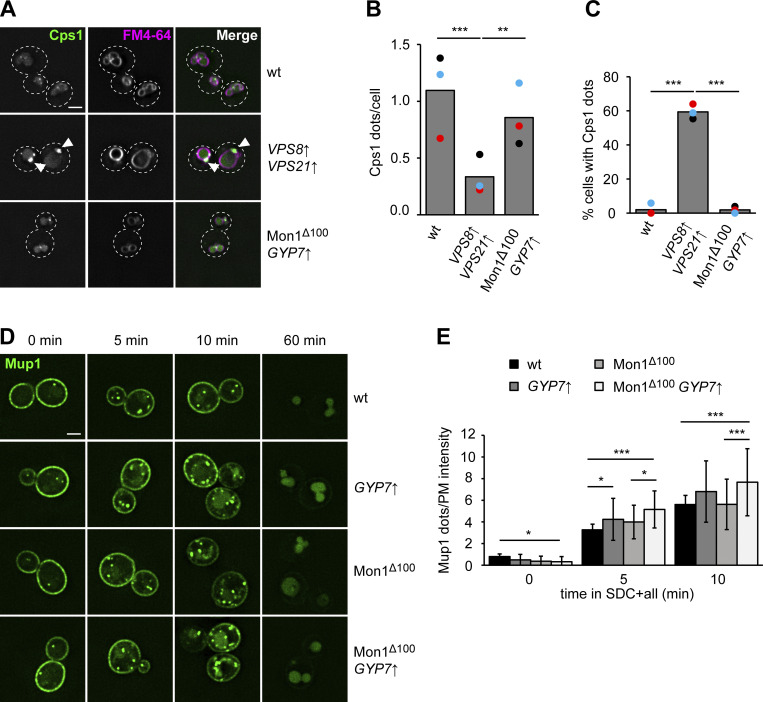
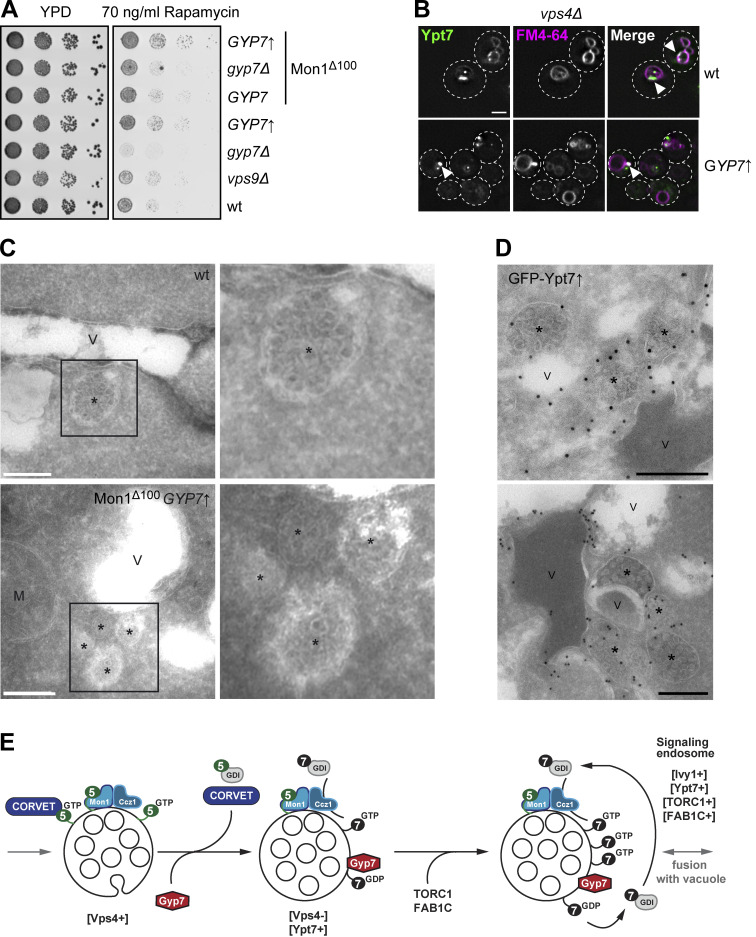
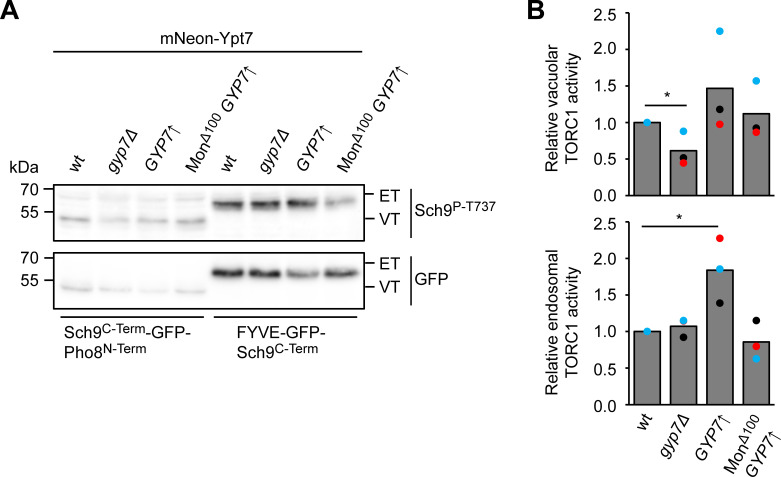
Organelles of the endomembrane system contain Rab GTPases as identity markers. Their localization is determined by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs). It remains largely unclear how these regulators are specifically targeted to organelles and how their activity is regulated. Here, we focus on the GAP Gyp7, which acts on the Rab7-like Ypt7 protein in yeast, and surprisingly observe the protein exclusively in puncta proximal to the vacuole. Mistargeting of Gyp7 to the vacuole strongly affects vacuole morphology, suggesting that endosomal localization is needed for function. In agreement, efficient endolysosomal transport requires Gyp7. In vitro assays reveal that Gyp7 requires a distinct lipid environment for membrane binding and activity. Overexpression of Gyp7 concentrates Ypt7 in late endosomes and results in resistance to rapamycin, an inhibitor of the target of rapamycin complex 1 (TORC1), suggesting that these late endosomes are signaling endosomes. We postulate that Gyp7 is part of regulatory machinery involved in late endosome function.
© 2024 Füllbrunn et al.
Conflict of interest statement
Disclosures: The authors declare no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
