

3D molecular generative framework for interaction-guided drug design
- PMID: 38538598
- PMCID: PMC10973397
- DOI: 10.1038/s41467-024-47011-2
3D molecular generative framework for interaction-guided drug design
Abstract
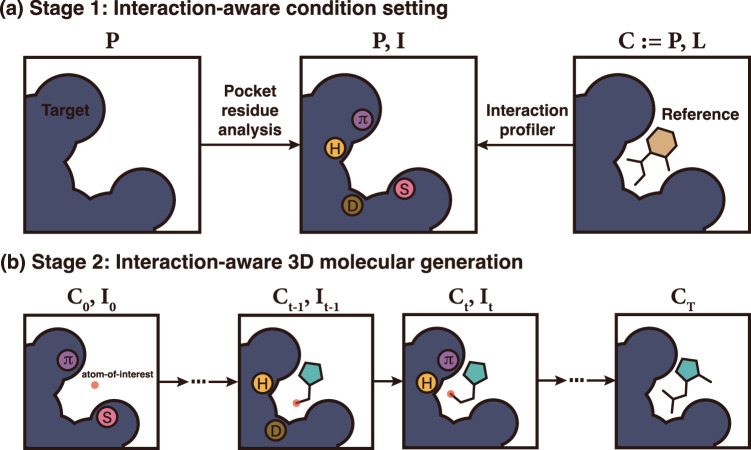
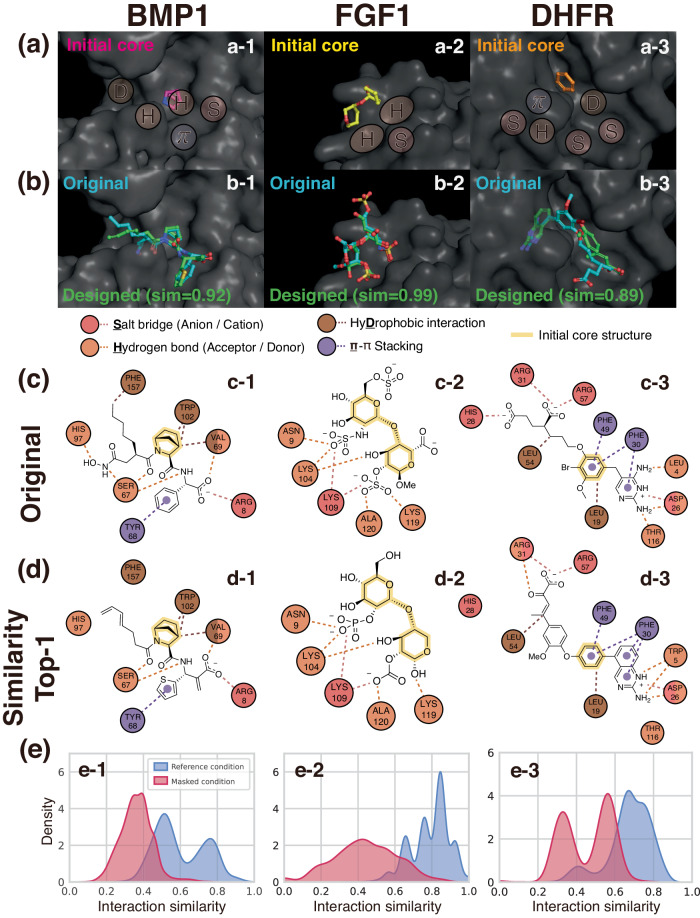
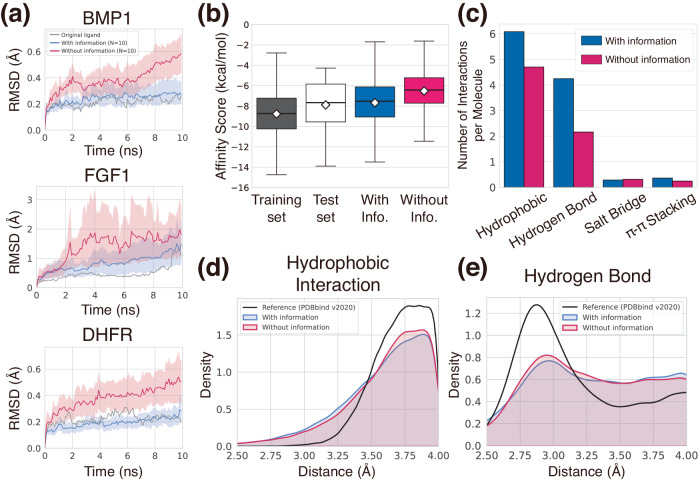
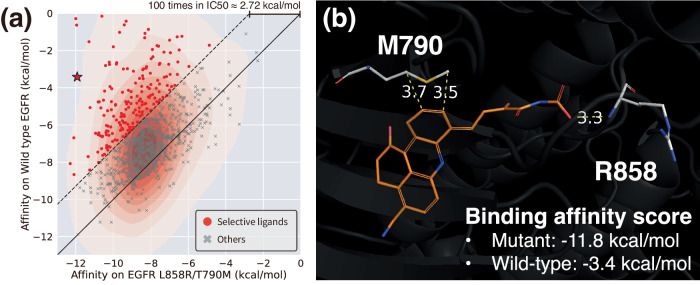
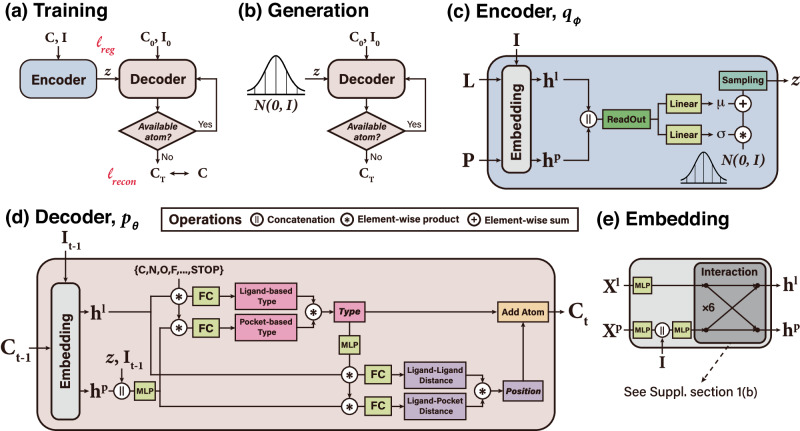
Deep generative modeling has a strong potential to accelerate drug design. However, existing generative models often face challenges in generalization due to limited data, leading to less innovative designs with often unfavorable interactions for unseen target proteins. To address these issues, we propose an interaction-aware 3D molecular generative framework that enables interaction-guided drug design inside target binding pockets. By leveraging universal patterns of protein-ligand interactions as prior knowledge, our model can achieve high generalizability with limited experimental data. Its performance has been comprehensively assessed by analyzing generated ligands for unseen targets in terms of binding pose stability, affinity, geometric patterns, diversity, and novelty. Moreover, the effective design of potential mutant-selective inhibitors demonstrates the applicability of our approach to structure-based drug design.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Muralidhar, N., Islam, M., Marwah, M., Karpatne, A. & Ramakrishnan, N., Incorporating prior domain knowledge into deep neural networks. In: 2018 IEEE International Conference On Big Data (big Data) 36–45 (IEEE, 2018).
-
- Yu Y, et al. Techniques and challenges of image segmentation: a review. Electronics. 2023;12:1199. doi: 10.3390/electronics12051199. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
