

Exploring the Role of Platelets in Virus-Induced Inflammatory Demyelinating Disease and Myocarditis
- PMID: 38542433
- PMCID: PMC10970283
- DOI: 10.3390/ijms25063460
Exploring the Role of Platelets in Virus-Induced Inflammatory Demyelinating Disease and Myocarditis
Abstract
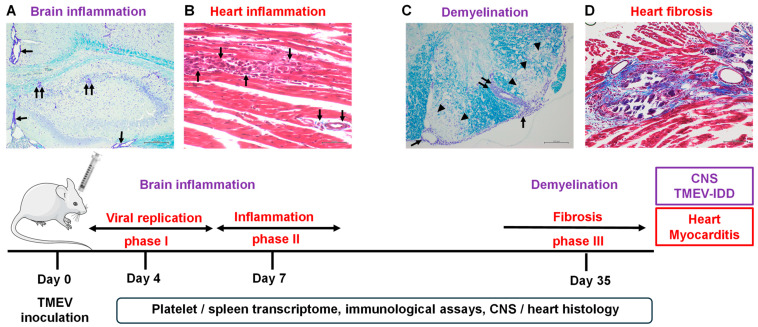
Theiler's murine encephalomyelitis virus (TMEV) infection has been used as a mouse model for two virus-induced organ-specific immune-mediated diseases. TMEV-induced demyelinating disease (TMEV-IDD) in the central nervous system (CNS) is a chronic inflammatory disease with viral persistence and an animal model of multiple sclerosis (MS) in humans. TMEV infection can also cause acute myocarditis with viral replication and immune cell infiltration in the heart, leading to cardiac fibrosis. Since platelets have been reported to modulate immune responses, we aimed to determine the role of platelets in TMEV infection. In transcriptome analyses of platelets, distinct sets of immune-related genes, including major histocompatibility complex (MHC) class I, were up- or downregulated in TMEV-infected mice at different time points. We depleted platelets from TMEV-infected mice by injecting them with platelet-specific antibodies. The platelet-depleted mice had significantly fewer viral antigen-positive cells in the CNS. Platelet depletion reduced the severities of TMEV-IDD and myocarditis, although the pathology scores did not reach statistical significance. Immunologically, the platelet-depleted mice had an increase in interferon (IFN)-γ production with a higher anti-TMEV IgG2a/IgG1 ratio. Thus, platelets may play roles in TMEV infection, such as gene expression, viral clearance, and anti-viral antibody isotype responses.
Keywords: RNA sequencing analyses; bioinformatics analysis; dilated cardiomyopathy; glycoprotein Ib α chain; neuroinflammatory disease; picornavirus infections; regulation of gene expression.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures









Similar articles
-
Theiler's Virus-Mediated Immunopathology in the CNS and Heart: Roles of Organ-Specific Cytokine and Lymphatic Responses.Front Immunol. 2018 Dec 10;9:2870. doi: 10.3389/fimmu.2018.02870. eCollection 2018. Front Immunol. 2018. PMID: 30619258 Free PMC article.
-
CNS expression of B7-H1 regulates pro-inflammatory cytokine production and alters severity of Theiler's virus-induced demyelinating disease.PLoS One. 2011 Apr 8;6(4):e18548. doi: 10.1371/journal.pone.0018548. PLoS One. 2011. PMID: 21494618 Free PMC article.
-
Coexpression of class I major histocompatibility antigen and viral RNA in central nervous system of mice infected with Theiler's virus: a model for multiple sclerosis.Mayo Clin Proc. 1992 Sep;67(9):829-38. doi: 10.1016/s0025-6196(12)60820-9. Mayo Clin Proc. 1992. PMID: 1434926
-
Excessive Innate Immunity Steers Pathogenic Adaptive Immunity in the Development of Theiler's Virus-Induced Demyelinating Disease.Int J Mol Sci. 2021 May 17;22(10):5254. doi: 10.3390/ijms22105254. Int J Mol Sci. 2021. PMID: 34067536 Free PMC article. Review.
-
Facets of Theiler's Murine Encephalomyelitis Virus-Induced Diseases: An Update.Int J Mol Sci. 2019 Jan 21;20(2):448. doi: 10.3390/ijms20020448. Int J Mol Sci. 2019. PMID: 30669615 Free PMC article. Review.
Cited by
-
A New Perspective on Mechanisms of Neurodegeneration in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis: the Early and Critical Role of Platelets in Neuro/Axonal Loss.J Neuroimmune Pharmacol. 2025 Feb 4;20(1):14. doi: 10.1007/s11481-025-10182-w. J Neuroimmune Pharmacol. 2025. PMID: 39904925 Free PMC article. Review.
-
Gut Microbiota in a Viral Model of Multiple Sclerosis: Modulation and Pitfalls by Oral Antibiotic Treatment.Cells. 2025 Jun 9;14(12):871. doi: 10.3390/cells14120871. Cells. 2025. PMID: 40558498 Free PMC article.
References
-
- Flint S.J., Racaniello V.R., Rall G.F., Hatziioannou T., Skalka A.M. Patterns and pathogenesis. In: Flint S.J., Racaniello V.R., Rall G.F., Hatziioannou T., Skalka A.M., editors. Principles of Virology, Volume 2: Pathogenesis and Control. 5th ed. ASM Press; Washington, DC, USA: 2020. pp. 146–186.
MeSH terms
Substances
Grants and funding
- (RSWF\R3\183001) (F.N.E.G.)/The Royal Society Wolfson Foundation
- (Orange funding) (H.T.)/The Hokkaido Lawyer Association for hepatitis B
- (A seeds) (H.T.)/Hokkaido Organization for Translational Research
- MEXT, 2021-2025, scholarship (I.A.)/Ministry of Education, Culture, Sports, Science and Technology
- JP22K07527 (S.O.), JP23K06493 (F.S.), JP21K07167 (H.T.), JP22K18378 and JP23K08901 (I.T.)/Japan Society for the Promotion of Science
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
