

Genome and RNA sequencing boost neuromuscular diagnoses to 62% from 34% with exome sequencing alone
- PMID: 38544359
- PMCID: PMC11093248
- DOI: 10.1002/acn3.52041
Genome and RNA sequencing boost neuromuscular diagnoses to 62% from 34% with exome sequencing alone
Abstract
Objective: Most families with heritable neuromuscular disorders do not receive a molecular diagnosis. Here we evaluate diagnostic utility of exome, genome, RNA sequencing, and protein studies and provide evidence-based recommendations for their integration into practice.
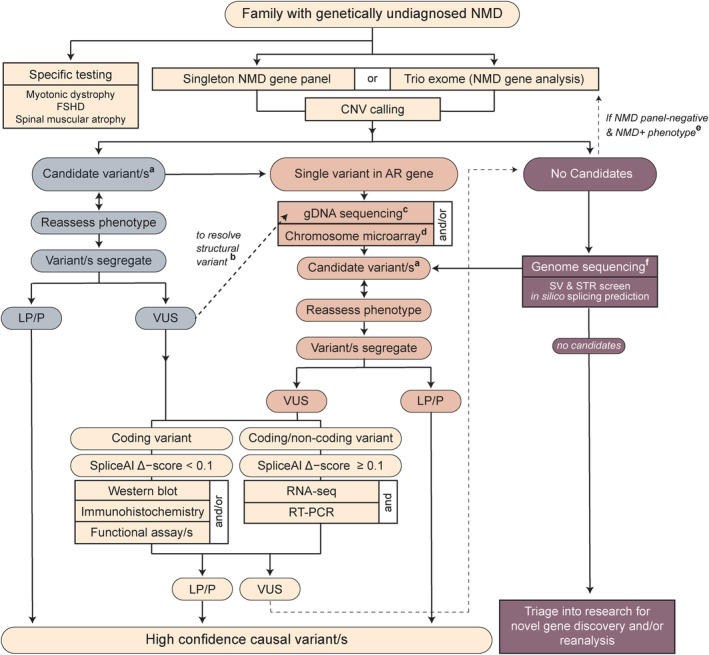
Methods: In total, 247 families with suspected monogenic neuromuscular disorders who remained without a genetic diagnosis after standard diagnostic investigations underwent research-led massively parallel sequencing: neuromuscular disorder gene panel, exome, genome, and/or RNA sequencing to identify causal variants. Protein and RNA studies were also deployed when required.
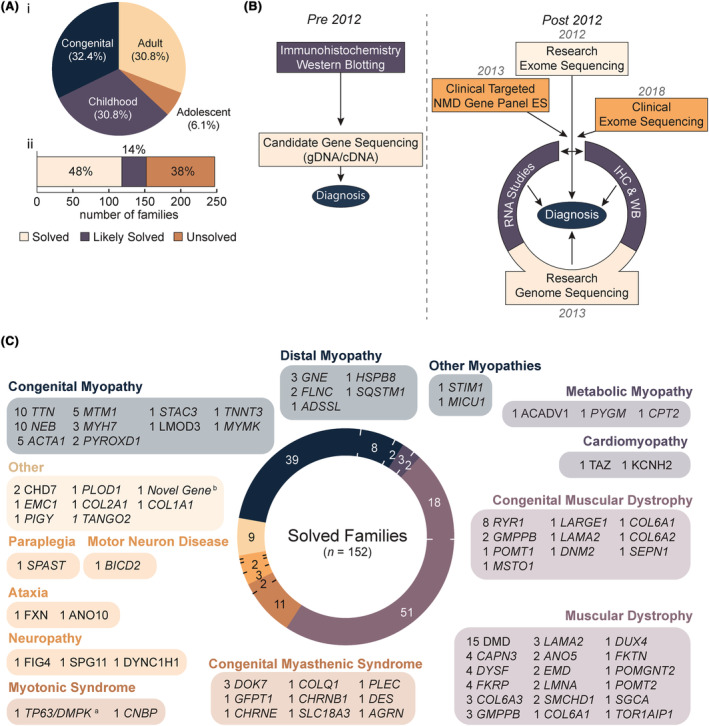
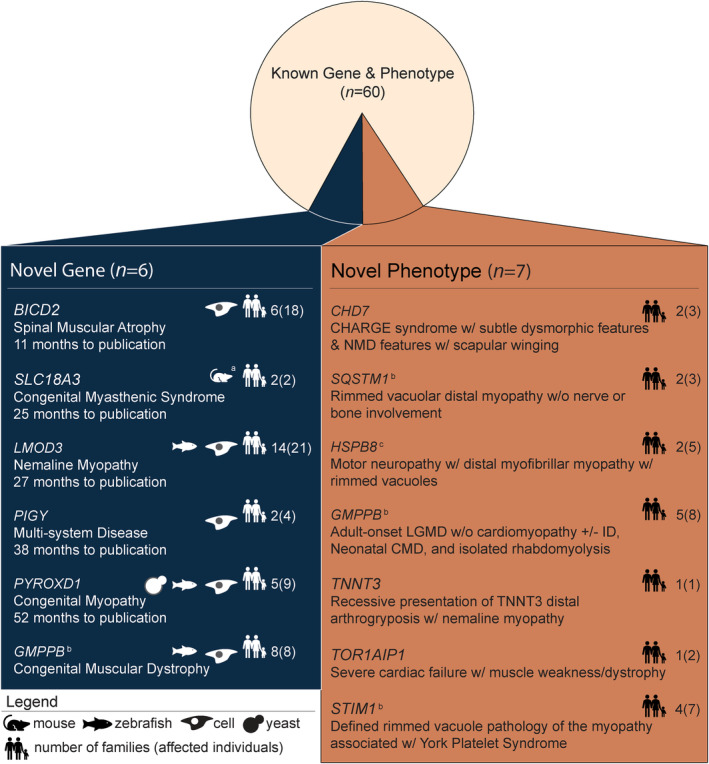
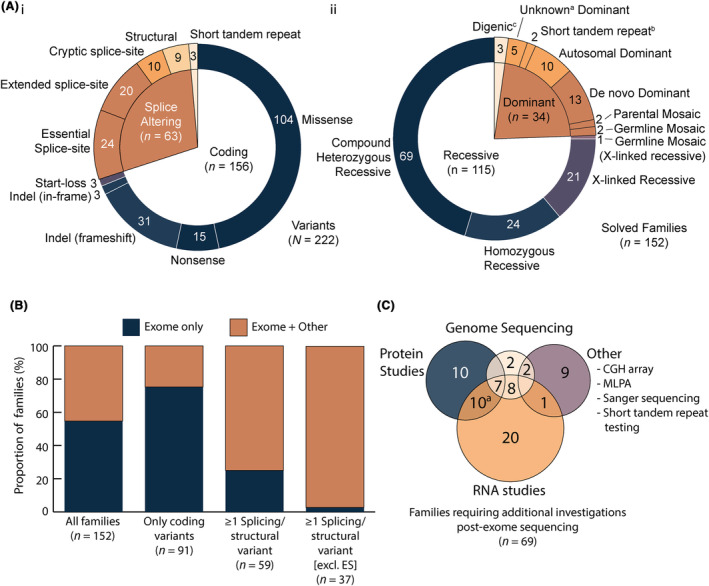
Results: Integration of exome sequencing and auxiliary genome, RNA and/or protein studies identified causal or likely causal variants in 62% (152 out of 247) of families. Exome sequencing alone informed 55% (83 out of 152) of diagnoses, with remaining diagnoses (45%; 69 out of 152) requiring genome sequencing, RNA and/or protein studies to identify variants and/or support pathogenicity. Arrestingly, novel disease genes accounted for <4% (6 out of 152) of diagnoses while 36.2% of solved families (55 out of 152) harbored at least one splice-altering or structural variant in a known neuromuscular disorder gene. We posit that contemporary neuromuscular disorder gene-panel sequencing could likely provide 66% (100 out of 152) of our diagnoses today.
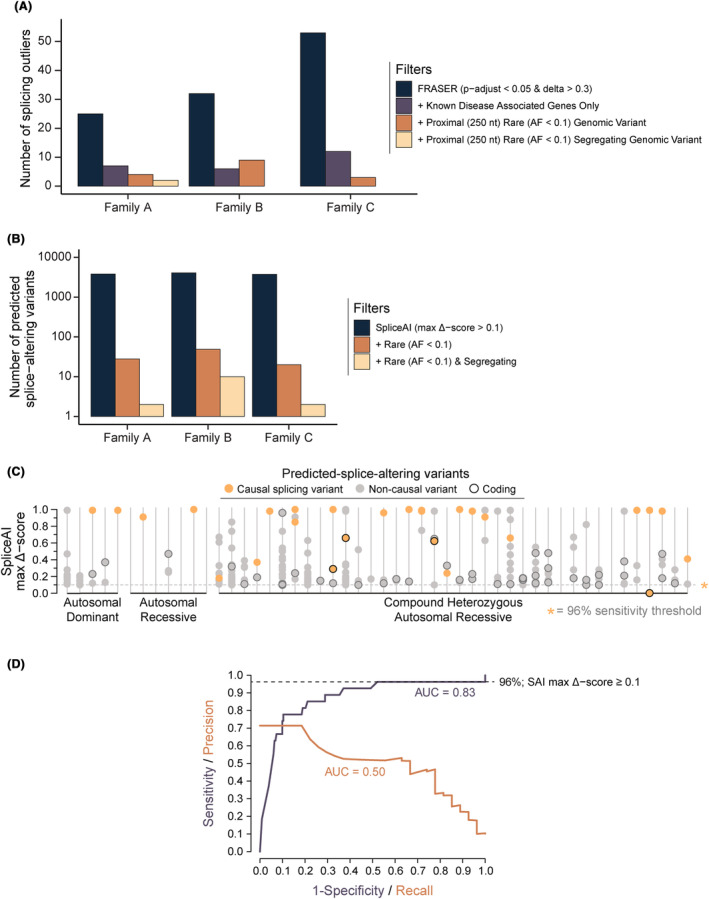
Interpretation: Our results emphasize thorough clinical phenotyping to enable deep scrutiny of all rare genetic variation in phenotypically consistent genes. Post-exome auxiliary investigations extended our diagnostic yield by 81% overall (34-62%). We present a diagnostic algorithm that details deployment of genomic and auxiliary investigations to obtain these diagnoses today most effectively. We hope this provides a practical guide for clinicians as they gain greater access to clinical genome and transcriptome sequencing.
© 2024 The Authors. Annals of Clinical and Translational Neurology published by Wiley Periodicals LLC on behalf of American Neurological Association.
Conflict of interest statement
S.T. Cooper is director of Frontier Genomics Pty Ltd (Australia). S.T. Cooper receives no remuneration (salary or consultancy fees) for this role. Frontier Genomics Pty Ltd has no current financial interests that will benefit from publication of this data. S.T. Cooper is a named inventor on intellectual property owned jointly by the University of Sydney and Sydney Children's Hospitals Network. This IP relates to splicing variant detection and interpretation and is licensed by Frontier Genomics Pty Ltd. S.T. Cooper is named inventor on Australian Patent No. 2019379868 and Australian Provisional Patent No. 2019900836. The remaining co‐authors declare no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
