

Clinical and Genetic Analysis of Patients With TK2 Deficiency
- PMID: 38544965
- PMCID: PMC10965359
- DOI: 10.1212/NXG.0000000000200138
Clinical and Genetic Analysis of Patients With TK2 Deficiency
Abstract
Objectives: Thymidine kinase 2 deficiency (TK2d) is a rare autosomal recessive disorder that stems from a perturbation of the mitochondrial DNA maintenance. Nucleoside treatment has recently shown promise as a disease-modifying therapy. TK2d was initially associated with rapidly progressive fatal myopathy in children featuring mitochondrial DNA depletion. Subsequently, less severe variants of the disease were described, with onset of symptoms during adolescence or adulthood and associated with the presence of multiple mtDNA deletions. These less severe phenotypes have been reported in only 15% of the approximately 120 patients described worldwide. However, some reports suggest that these juvenile and adult-onset presentations may be more common. The objective of this study was to describe the clinical phenotype in a sample of patients from Spain.
Methods: This study includes 53 patients harboring biallelic TK2 pathogenic variants, compiling data retrospectively from 7 Spanish centers. We analyzed allele frequency, investigated the most recent common ancestor of core haplotypes, and used the Runs of Homozygosity approach to investigate variant coalescence.
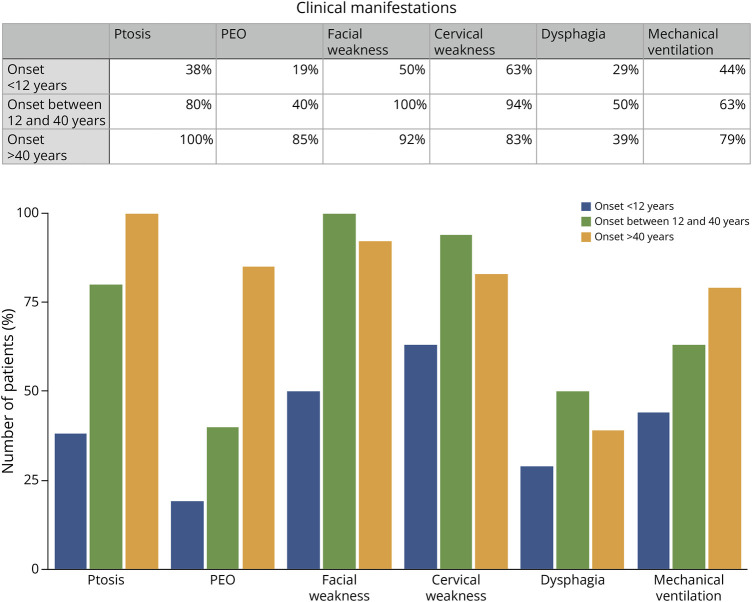
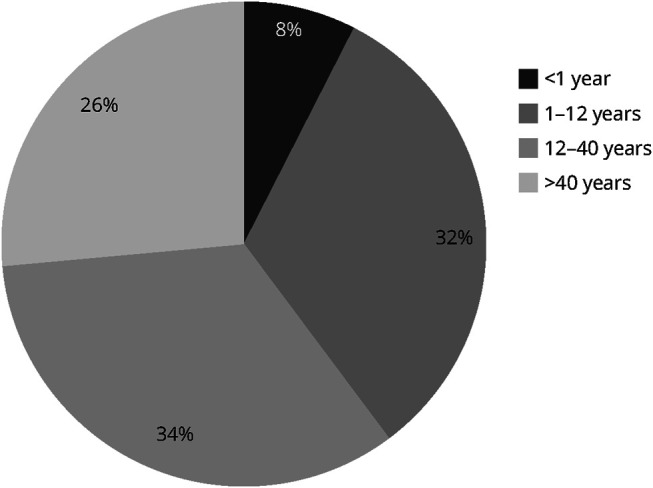
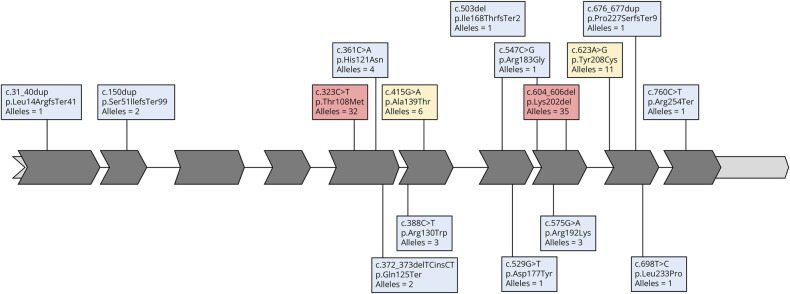
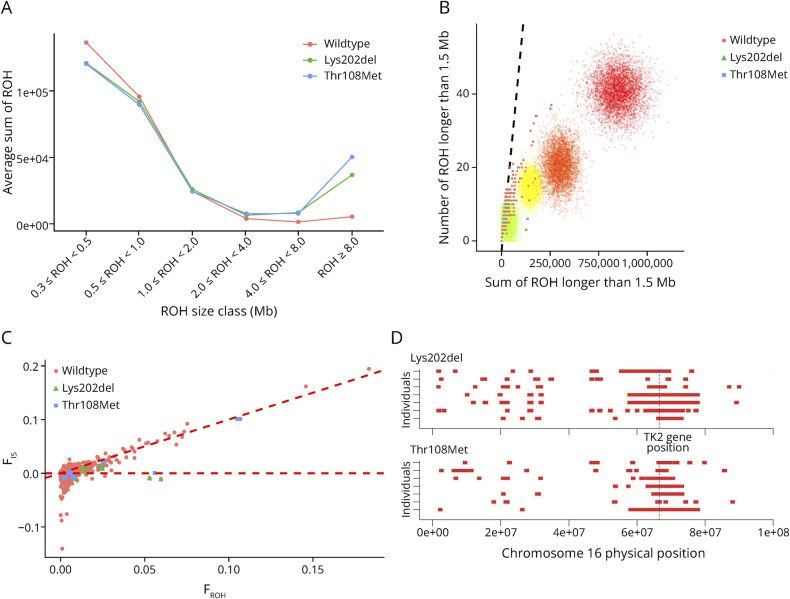
Results: Symptom onset distribution revealed that 32 patients (60%) experienced symptoms beyond 12 years of age. Approximately 30% of patients died of respiratory insufficiency, while 56% of surviving patients needed mechanical ventilation. Genetic analysis identified 16 distinct variants in TK2. Two variants, p.Lys202del and p.Thr108Met, exhibited significantly higher prevalence in the Spanish population than that reported in gnomAD database (86-fold and 13-fold, respectively). These variants are estimated to have originated approximately 16.8 generations ago for p.Thr108Met and 95.2 generations ago for p.Lys202del within the Spanish population, with the increase in frequency attributed to various forms of inbreeding. In late-onset cases, 46.9% carried the p.Lys202del variant.
Discussion: The higher frequency of TK2d in Spain can be partially attributed to the increased prevalence of 2 variants and consanguinity. Notably, in 60% of the cohort, the disease was late-onset, emphasizing the potential underdiagnosis of this subgroup of patients in other regions. Raising awareness of this potentially treatable disorder is of utmost importance because early interventions can significantly affect the quality of life and survival of affected individuals.
Copyright © 2024 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Conflict of interest statement
F. Ceballos reports no disclosures relevant to the manuscript, P. Serrano-Lorenzo reports no disclosures relevant to the manuscript, L. Bermejo-Guerrero reports no disclosures relevant to the manuscript, A. Blázquez reports no disclosures relevant to the manuscript, J.F. Quesada-Espinosa reports no disclosures relevant to the manuscript, J. Amigo reports no disclosures relevant to the manuscript, P. Minguez reports no disclosures relevant to the manuscript, C. Ayuso reports no disclosures relevant to the manuscript, E. García-Arumí reports no disclosures relevant to the manuscript, N. Muelas reports no disclosures relevant to the manuscript, T. Jaijo reports no disclosures relevant to the manuscript; A. Nascimento serves on the advisory board of UCB Pharma; B. Galán-Rodriguez reports no disclosures relevant to the manuscript; C. Paradas serves on the advisory board of UCB Pharma; TK2d Spanish-Group report no disclosures relevant to the manuscript; J. Arenas reports no disclosures relevant to the manuscript; A. Carracedo reports no disclosures relevant to the manuscript; R. Martí serves on the advisory board of UCB Pharma; MAM serves on the advisory board of UCB Pharma; and C. Domínguez-González serves on the advisory board of UCB Pharma. Go to Neurology.org/NG for full disclosures.
Figures
References
LinkOut - more resources
Full Text Sources