

Mitochondrial injury induced by a Salmonella genotoxin triggers the proinflammatory senescence-associated secretory phenotype
- PMID: 38555361
- PMCID: PMC10981749
- DOI: 10.1038/s41467-024-47190-y
Mitochondrial injury induced by a Salmonella genotoxin triggers the proinflammatory senescence-associated secretory phenotype
Abstract
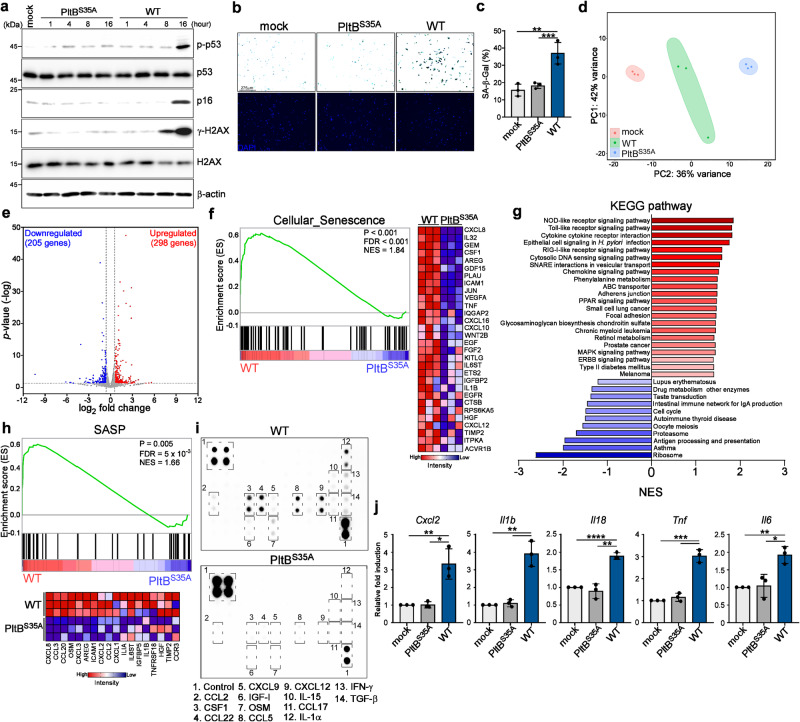
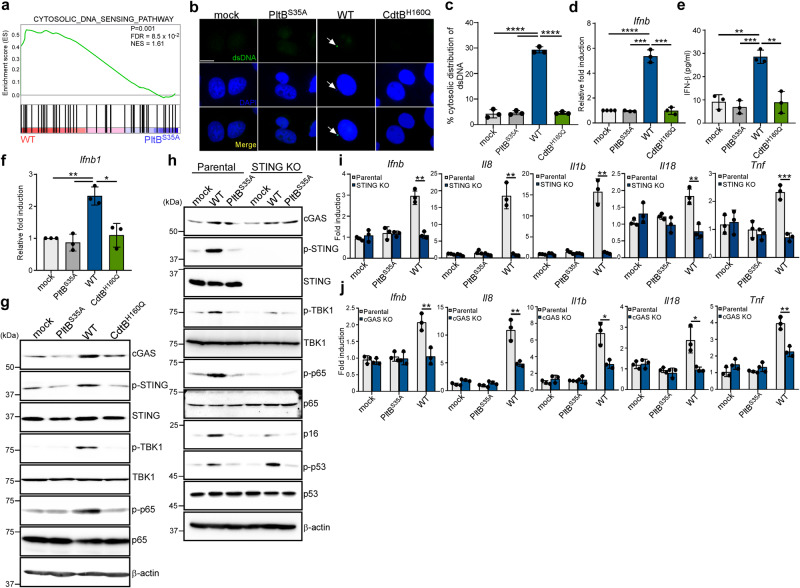
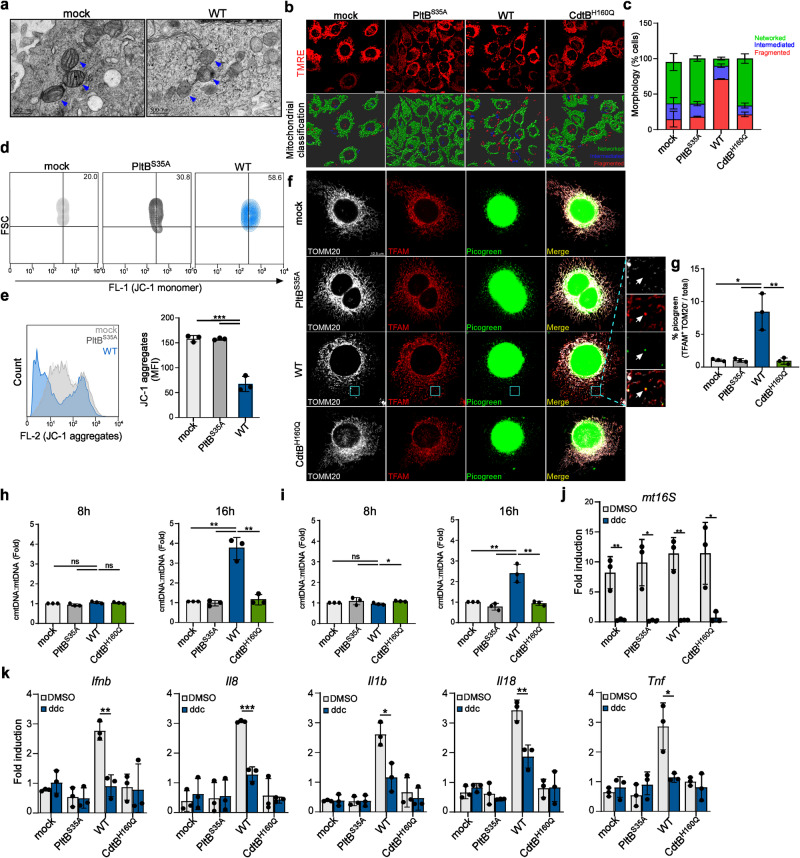
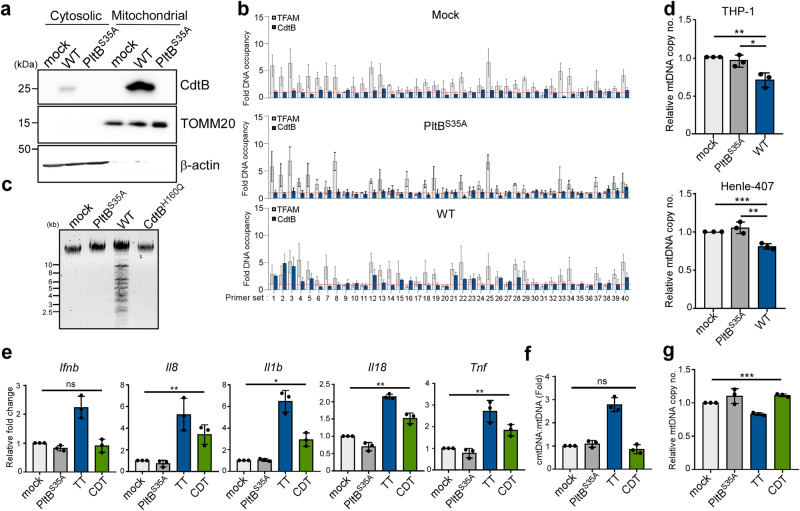
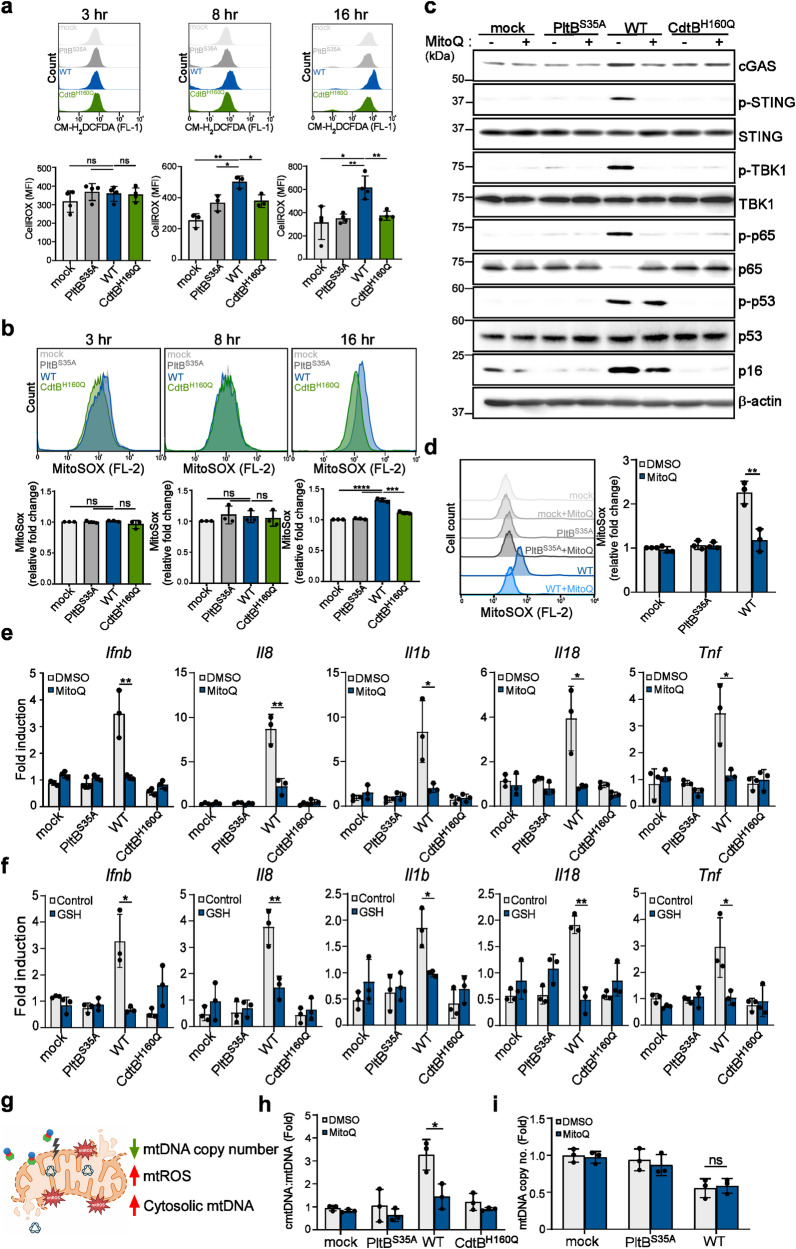
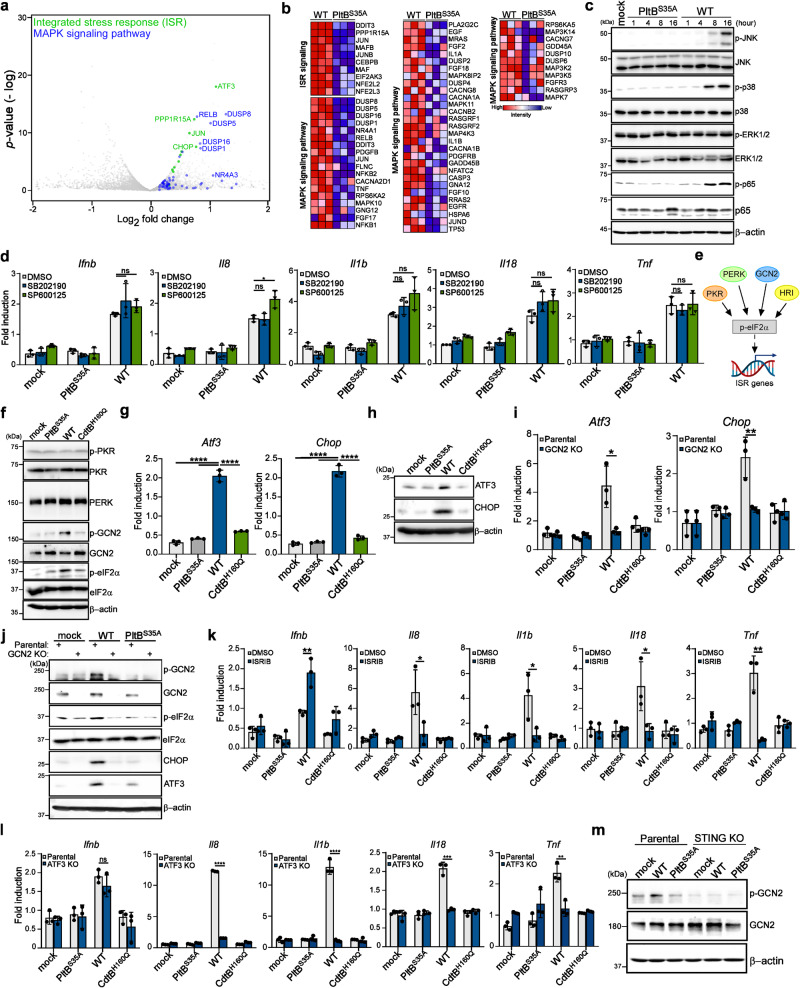
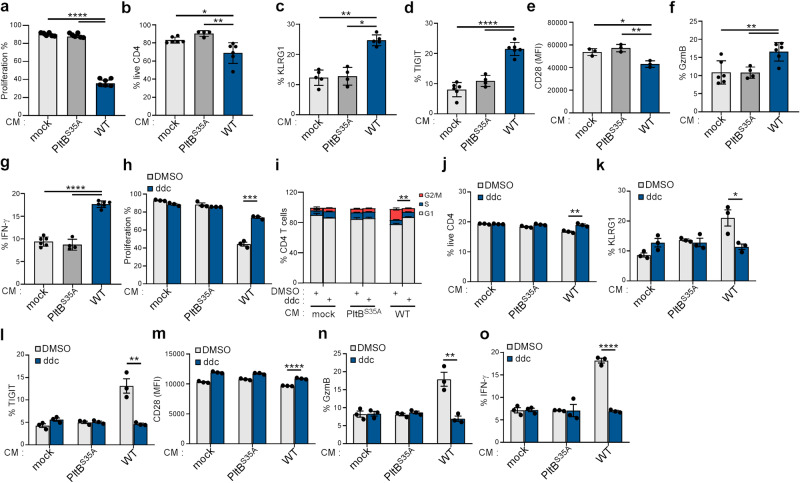
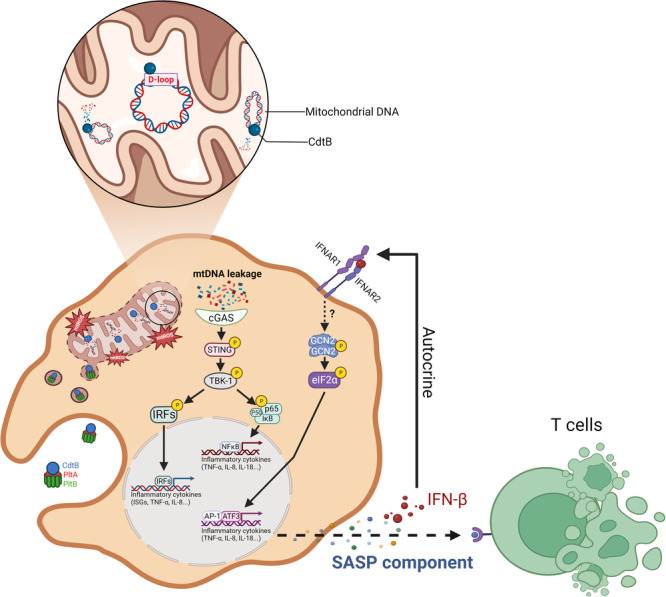
Bacterial genotoxins damage host cells by targeting their chromosomal DNA. In the present study, we demonstrate that a genotoxin of Salmonella Typhi, typhoid toxin, triggers the senescence-associated secretory phenotype (SASP) by damaging mitochondrial DNA. The actions of typhoid toxin disrupt mitochondrial DNA integrity, leading to mitochondrial dysfunction and disturbance of redox homeostasis. Consequently, it facilitates the release of damaged mitochondrial DNA into the cytosol, activating type I interferon via the cGAS-STING pathway. We also reveal that the GCN2-mediated integrated stress response plays a role in the upregulation of inflammatory components depending on the STING signaling axis. These SASP factors can propagate the senescence effect on T cells, leading to senescence in these cells. These findings provide insights into how a bacterial genotoxin targets mitochondria to trigger a proinflammatory SASP, highlighting a potential therapeutic target for an anti-toxin intervention.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
- MOST111-2636-B-002-028/Ministry of Science and Technology, Taiwan (Ministry of Science and Technology of Taiwan)
- NSTC112-2636-B-002-008/Ministry of Science and Technology, Taiwan (Ministry of Science and Technology of Taiwan)
- 112V1404-4/Ministry of Education (Ministry of Education, Republic of China (Taiwan))
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
