

SpatialcoGCN: deconvolution and spatial information-aware simulation of spatial transcriptomics data via deep graph co-embedding
- PMID: 38557675
- PMCID: PMC10982953
- DOI: 10.1093/bib/bbae130
SpatialcoGCN: deconvolution and spatial information-aware simulation of spatial transcriptomics data via deep graph co-embedding
Abstract
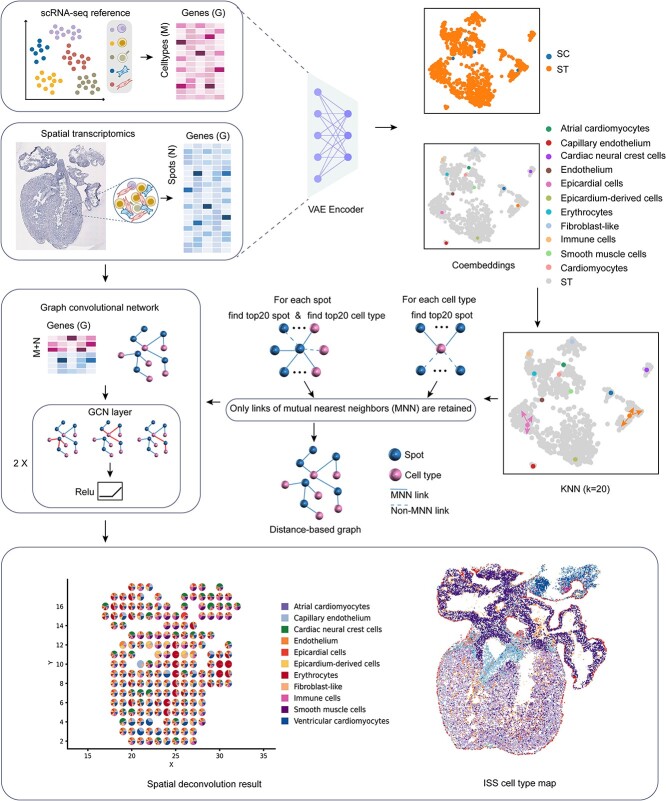
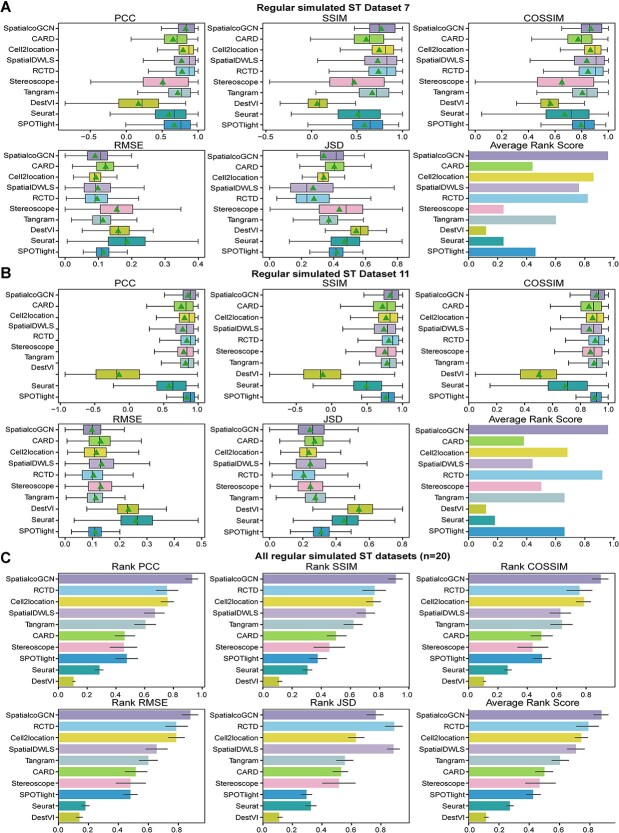
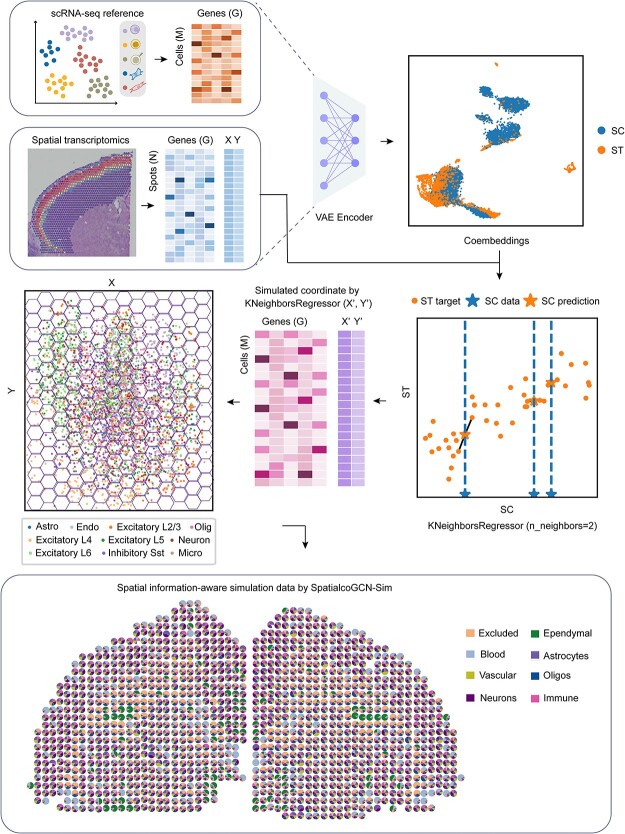
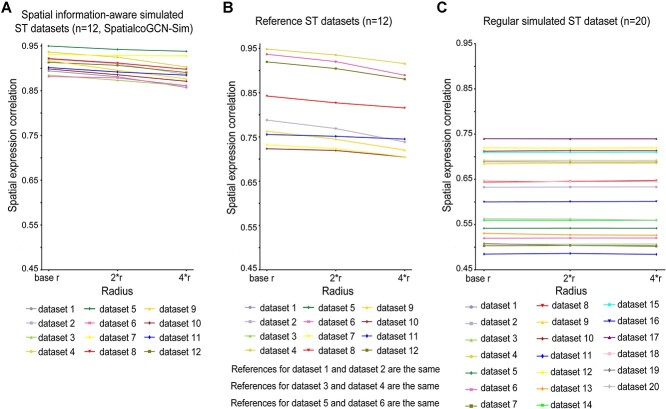
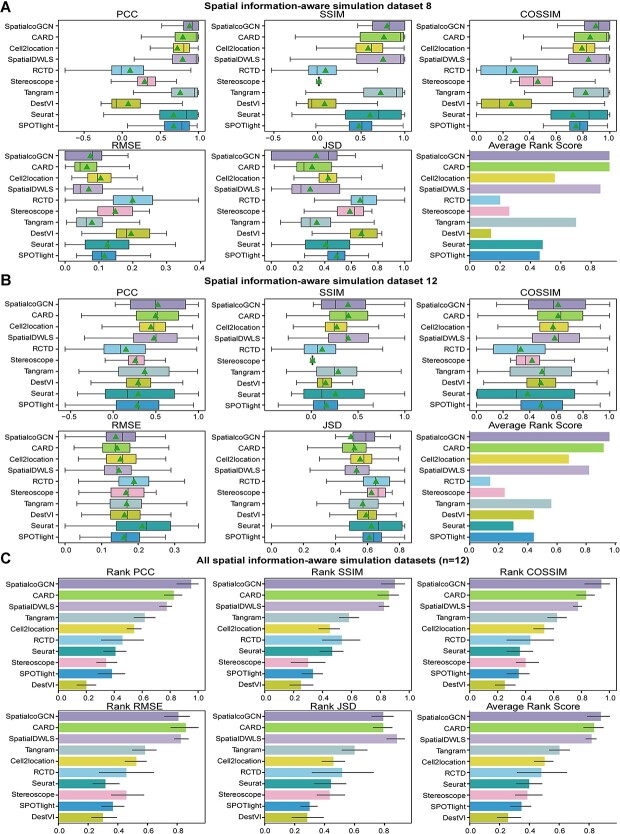
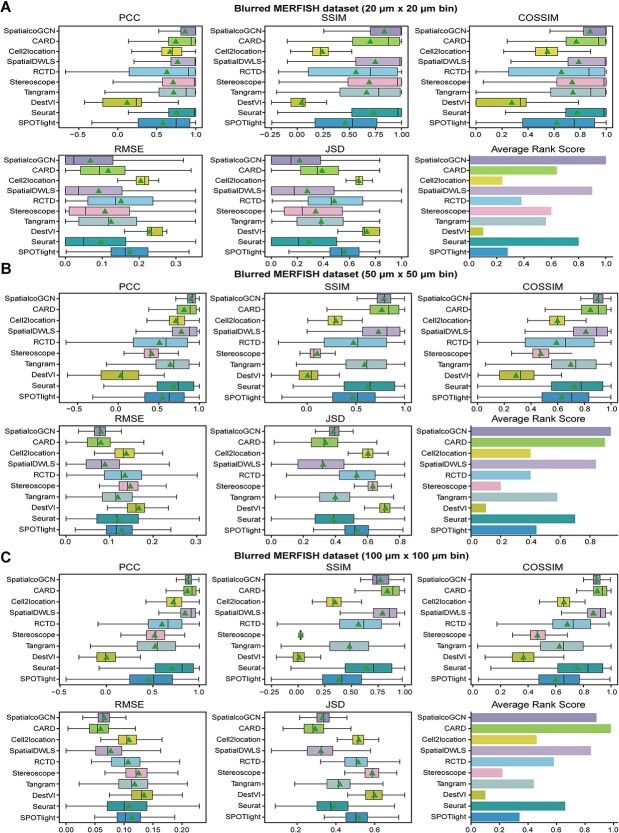
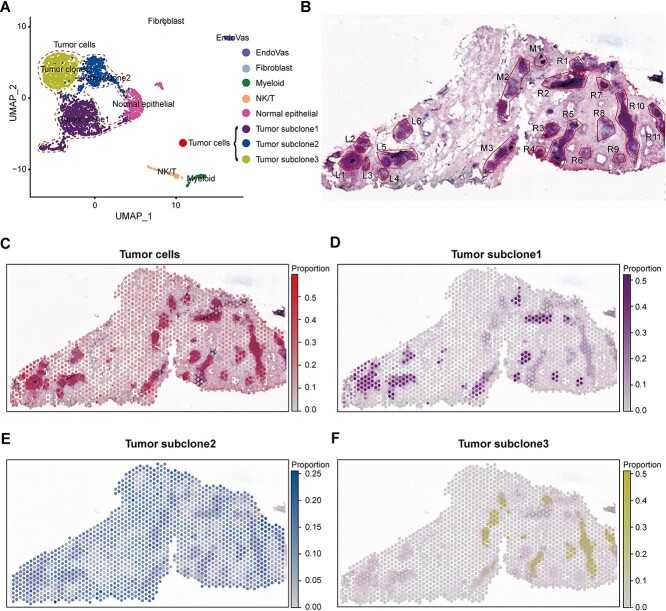
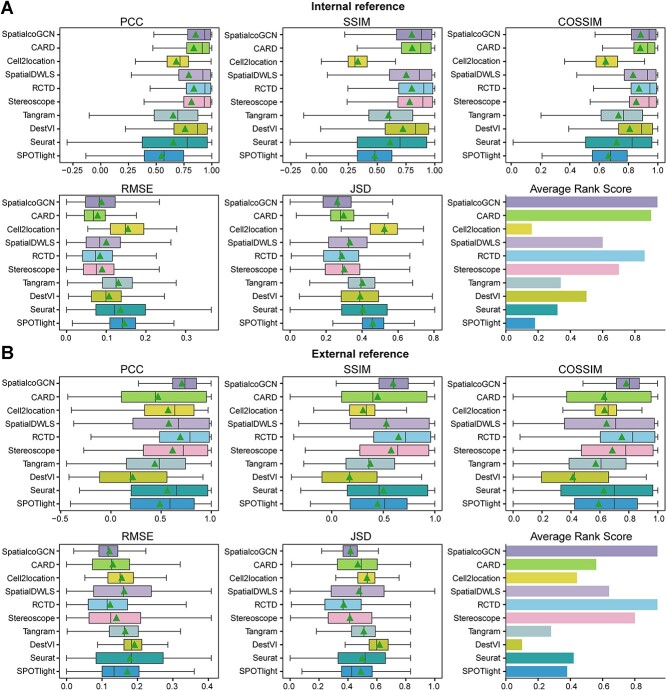
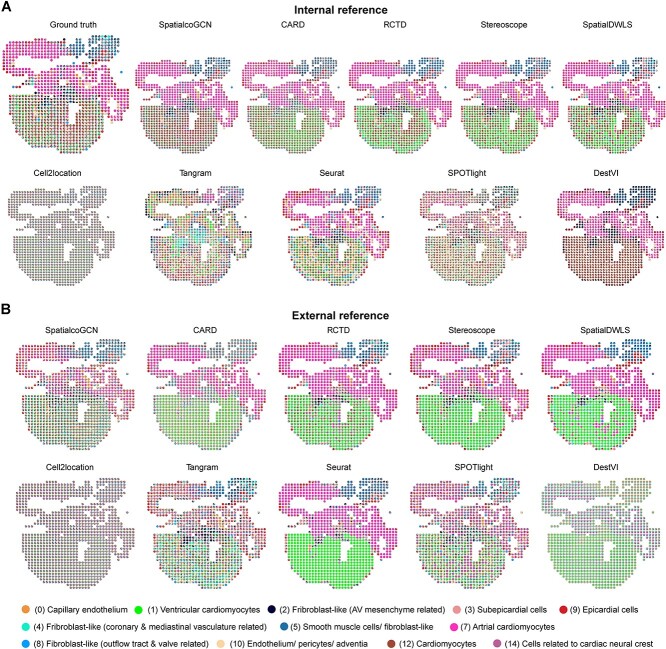
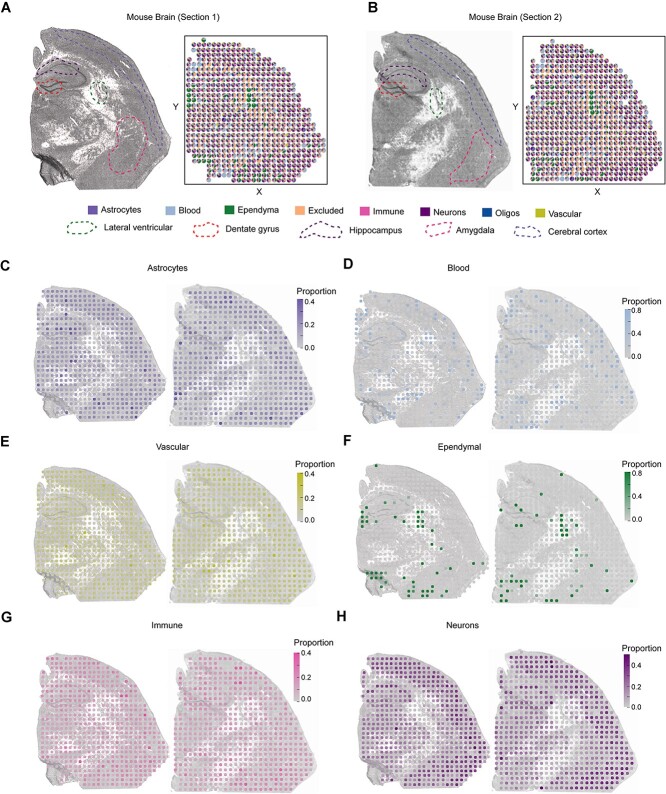
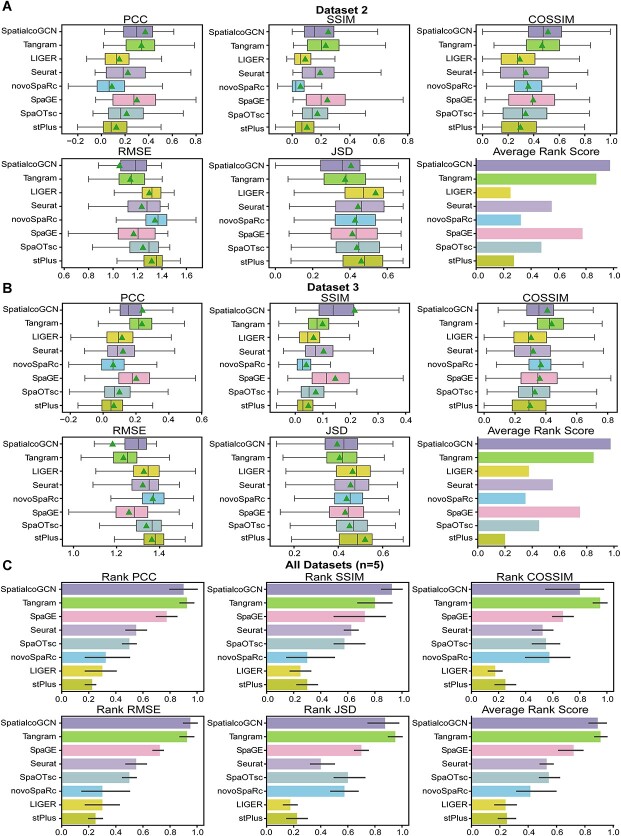
Spatial transcriptomics (ST) data have emerged as a pivotal approach to comprehending the function and interplay of cells within intricate tissues. Nonetheless, analyses of ST data are restricted by the low spatial resolution and limited number of ribonucleic acid transcripts that can be detected with several popular ST techniques. In this study, we propose that both of the above issues can be significantly improved by introducing a deep graph co-embedding framework. First, we establish a self-supervised, co-graph convolution network-based deep learning model termed SpatialcoGCN, which leverages single-cell data to deconvolve the cell mixtures in spatial data. Evaluations of SpatialcoGCN on a series of simulated ST data and real ST datasets from human ductal carcinoma in situ, developing human heart and mouse brain suggest that SpatialcoGCN could outperform other state-of-the-art cell type deconvolution methods in estimating per-spot cell composition. Moreover, with competitive accuracy, SpatialcoGCN could also recover the spatial distribution of transcripts that are not detected by raw ST data. With a similar co-embedding framework, we further established a spatial information-aware ST data simulation method, SpatialcoGCN-Sim. SpatialcoGCN-Sim could generate simulated ST data with high similarity to real datasets. Together, our approaches provide efficient tools for studying the spatial organization of heterogeneous cells within complex tissues.
Keywords: cell type deconvolution; graph-based deep learning; spatial transcriptomics; spatial transcriptomics data simulation.
© The Author(s) 2024. Published by Oxford University Press.
Figures

Similar articles
-
STCGAN: a novel cycle-consistent generative adversarial network for spatial transcriptomics cellular deconvolution.Brief Bioinform. 2024 Nov 22;26(1):bbae670. doi: 10.1093/bib/bbae670. Brief Bioinform. 2024. PMID: 39715685 Free PMC article.
-
STGAT: Graph attention networks for deconvolving spatial transcriptomics data.Comput Methods Programs Biomed. 2024 Dec;257:108431. doi: 10.1016/j.cmpb.2024.108431. Epub 2024 Sep 21. Comput Methods Programs Biomed. 2024. PMID: 39461117
-
Identifying spatial domains of spatially resolved transcriptomics via multi-view graph convolutional networks.Brief Bioinform. 2023 Sep 20;24(5):bbad278. doi: 10.1093/bib/bbad278. Brief Bioinform. 2023. PMID: 37544658
-
Computational solutions for spatial transcriptomics.Comput Struct Biotechnol J. 2022 Sep 1;20:4870-4884. doi: 10.1016/j.csbj.2022.08.043. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36147664 Free PMC article. Review.
-
A comprehensive comparison on cell-type composition inference for spatial transcriptomics data.Brief Bioinform. 2022 Jul 18;23(4):bbac245. doi: 10.1093/bib/bbac245. Brief Bioinform. 2022. PMID: 35753702 Free PMC article. Review.
Cited by
-
Cell-type deconvolution methods for spatial transcriptomics.Nat Rev Genet. 2025 May 14. doi: 10.1038/s41576-025-00845-y. Online ahead of print. Nat Rev Genet. 2025. PMID: 40369312 Review.
-
Accurate and Flexible Single Cell to Spatial Transcriptome Mapping with Celloc.Small Sci. 2024 Jun 26;4(10):2400139. doi: 10.1002/smsc.202400139. eCollection 2024 Oct. Small Sci. 2024. PMID: 40212250 Free PMC article.
-
Multi-task benchmarking of spatially resolved gene expression simulation models.Genome Biol. 2025 Mar 17;26(1):57. doi: 10.1186/s13059-025-03505-w. Genome Biol. 2025. PMID: 40098171 Free PMC article.
References
-
- Asp M, Bergenstråhle J, Lundeberg J. Spatially resolved transcriptomes—next generation tools for tissue exploration. Bioessays 2020;42:e1900221. - PubMed
-
- Stahl PL, Salmen F, Vickovic S, et al. . Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016;353:78–82. - PubMed
