

Multilevel superposition for deciphering the conformational variability of protein ensembles
- PMID: 38557679
- PMCID: PMC10983786
- DOI: 10.1093/bib/bbae137
Multilevel superposition for deciphering the conformational variability of protein ensembles
Abstract
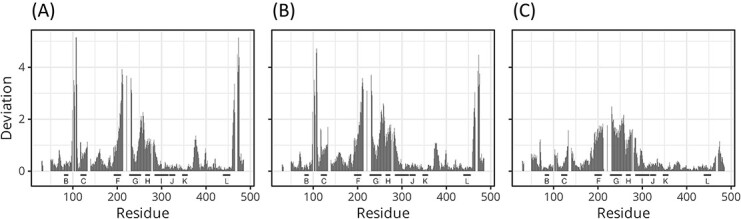
The dynamics and variability of protein conformations are directly linked to their functions. Many comparative studies of X-ray protein structures have been conducted to elucidate the relevant conformational changes, dynamics and heterogeneity. The rapid increase in the number of experimentally determined structures has made comparison an effective tool for investigating protein structures. For example, it is now possible to compare structural ensembles formed by enzyme species, variants or the type of ligands bound to them. In this study, the author developed a multilevel model for estimating two covariance matrices that represent inter- and intra-ensemble variability in the Cartesian coordinate space. Principal component analysis using the two estimated covariance matrices identified the inter-/intra-enzyme variabilities, which seemed to be important for the enzyme functions, with the illustrative examples of cytochrome P450 family 2 enzymes and class A $\beta$-lactamases. In P450, in which each enzyme has its own active site of a distinct size, an active-site motion shared universally between the enzymes was captured as the first principal mode of the intra-enzyme covariance matrix. In this case, the method was useful for understanding the conformational variability after adjusting for the differences between enzyme sizes. The developed method is advantageous in small ensemble-size problems and hence promising for use in comparative studies on experimentally determined structures where ensemble sizes are smaller than those generated, for example, by molecular dynamics simulations.
Keywords: EM algorithm; covariance matrix; principal component analysis; random effects model; structural superposition.
© The Author(s) 2024. Published by Oxford University Press.
Figures








References
-
- Kessel A, Ben-Tal N. Introduction to Proteins: Structure, Function, and Motion. Boca Raton: CRC Press, 2011. 10.1093/bib/bbad242, 10.1093/molbev/msab017. - DOI
-
- Campitelli P, Modi T, Kumar S, Ozkan SB. The role of conformational dynamics and Allostery in modulating protein evolution. Annu Rev Biophys 2020;49:267–88. - PubMed
