

This is a preprint.
Expanding the human gut microbiome atlas of Africa
- PMID: 38559015
- PMCID: PMC10980044
- DOI: 10.1101/2024.03.13.584859
Expanding the human gut microbiome atlas of Africa
Update in
-
Expanding the human gut microbiome atlas of Africa.Nature. 2025 Feb;638(8051):718-728. doi: 10.1038/s41586-024-08485-8. Epub 2025 Jan 29. Nature. 2025. PMID: 39880958 Free PMC article.
Abstract
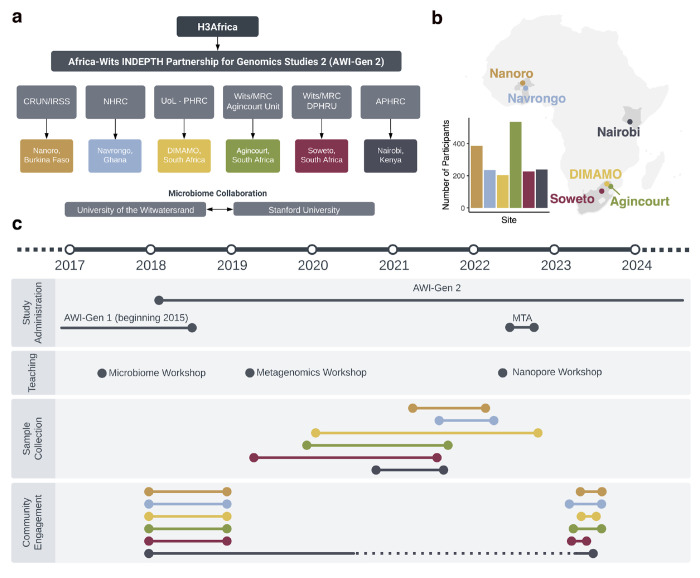
Population studies are crucial in understanding the complex interplay between the gut microbiome and geographical, lifestyle, genetic, and environmental factors. However, populations from low- and middle-income countries, which represent ~84% of the world population, have been excluded from large-scale gut microbiome research. Here, we present the AWI-Gen 2 Microbiome Project, a cross-sectional gut microbiome study sampling 1,803 women from Burkina Faso, Ghana, Kenya, and South Africa. By intensively engaging with communities that range from rural and horticultural to urban informal settlements and post-industrial, we capture population diversity that represents a far greater breadth of the world's population. Using shotgun metagenomic sequencing, we find that study site explains substantially more microbial variation than disease status. We identify taxa with strong geographic and lifestyle associations, including loss of Treponema and Cryptobacteroides species and gain of Bifidobacterium species in urban populations. We uncover a wealth of prokaryotic and viral novelty, including 1,005 new bacterial metagenome-assembled genomes, and identify phylogeography signatures in Treponema succinifaciens. Finally, we find a microbiome signature of HIV infection that is defined by several taxa not previously associated with HIV, including Dysosmobacter welbionis and Enterocloster sp. This study represents the largest population-representative survey of gut metagenomes of African individuals to date, and paired with extensive clinical biomarkers, demographic data, and lifestyle information, provides extensive opportunity for microbiome-related discovery and research.
Conflict of interest statement
Competing Interests The authors declare no competing interests.
Figures





References
-
- Gacesa R. et al. Environmental factors shaping the gut microbiome in a Dutch population. Nature 604, 732–739 (2022). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources