

This is a preprint.
RNA Polymerase II hypertranscription at histone genes in cancer FFPE samples
- PMID: 38559075
- PMCID: PMC10979862
- DOI: 10.1101/2024.02.28.582647
RNA Polymerase II hypertranscription at histone genes in cancer FFPE samples
Update in
-
RNA polymerase II at histone genes predicts outcome in human cancer.Science. 2025 Jan 2;387(6735):737-743. doi: 10.1126/science.ads2169. Epub 2025 Feb 13. Science. 2025. PMID: 39946483 Free PMC article.
Abstract
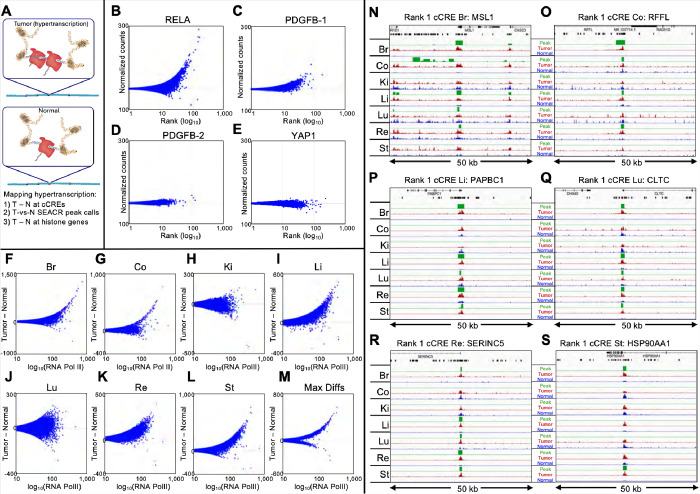
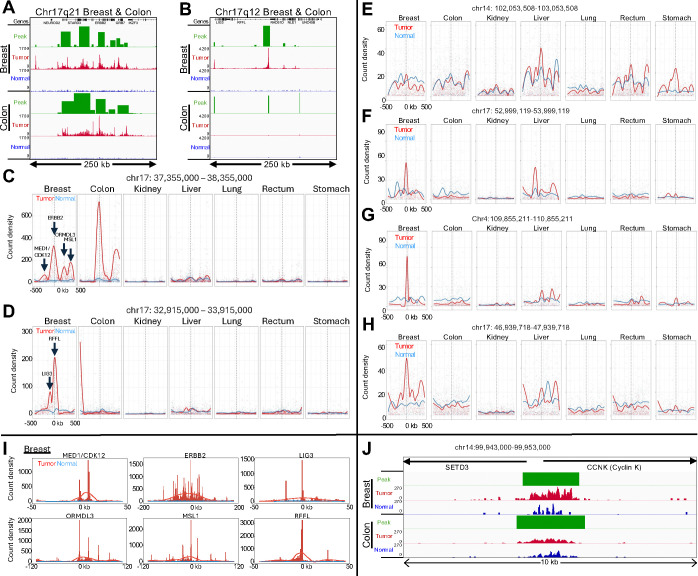
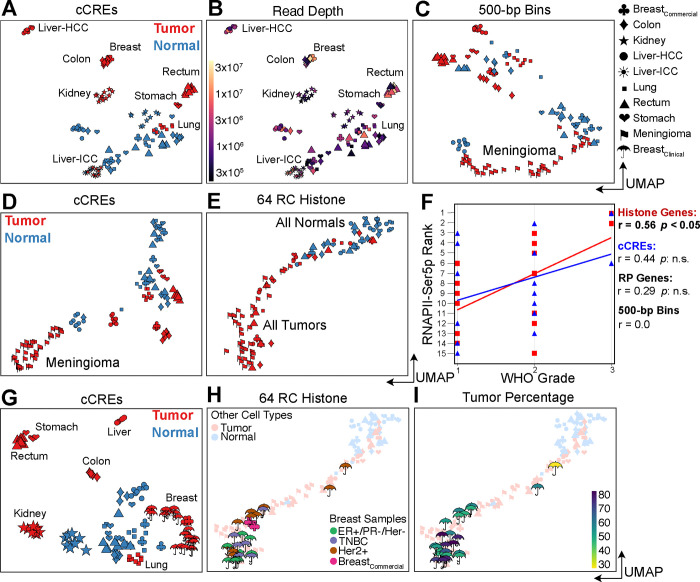
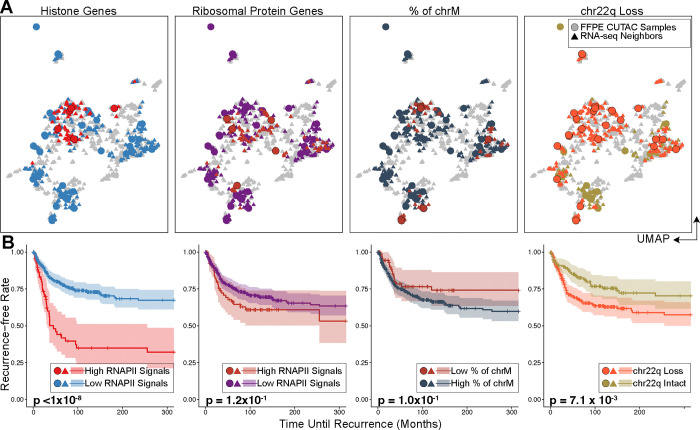
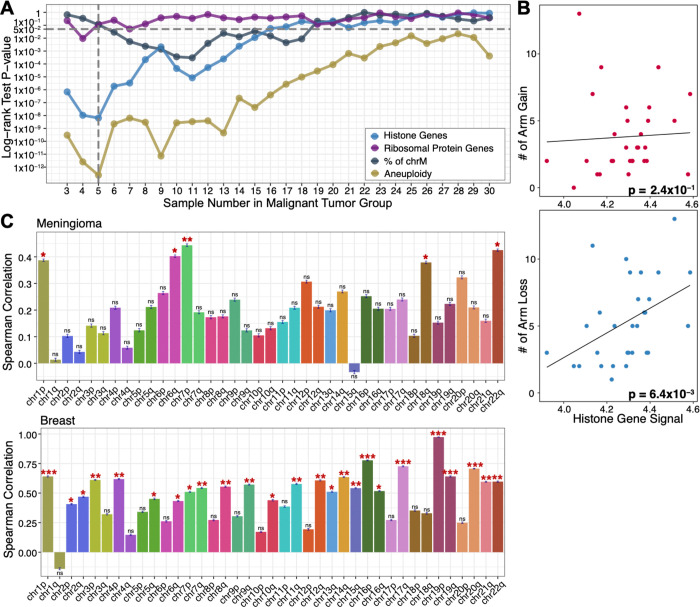
Genome-wide hypertranscription is common in human cancer and predicts poor prognosis. To understand how hypertranscription might drive cancer, we applied our FFPE-CUTAC method for mapping RNA Polymerase II (RNAPII) genome-wide in formalin-fixed paraffin-embedded (FFPE) sections. We demonstrate global RNAPII elevations in mouse gliomas and assorted human tumors in small clinical samples and discover regional elevations corresponding to de novo HER2 amplifications punctuated by likely selective sweeps. RNAPII occupancy at replication-coupled histone genes correlated with WHO grade in meningiomas, accurately predicted rapid recurrence, and corresponded to whole-arm chromosome losses. Elevated RNAPII at histone genes in meningiomas and diverse breast cancers is consistent with histone production being rate-limiting for S-phase progression and histone gene hypertranscription driving overproliferation and aneuploidy in cancer, with general implications for precision oncology.
Keywords: Centromeres; Epigenomics; Gene Regulation; HER2 amplification; Meningioma; Mitochondrial DNA; Whole-arm aneuploidy.
Conflict of interest statement
Competing interests S.H. is an inventor in a USPTO patent application filed by the Fred Hutchinson Cancer Center pertaining to CUTAC and FFPE-CUTAC (application number 63/505,964). The remaining authors declare no competing interests.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous