

This is a preprint.
Functional associations of evolutionarily recent human genes exhibit sensitivity to the 3D genome landscape and disease
- PMID: 38559085
- PMCID: PMC10980080
- DOI: 10.1101/2024.03.17.585403
Functional associations of evolutionarily recent human genes exhibit sensitivity to the 3D genome landscape and disease
Abstract
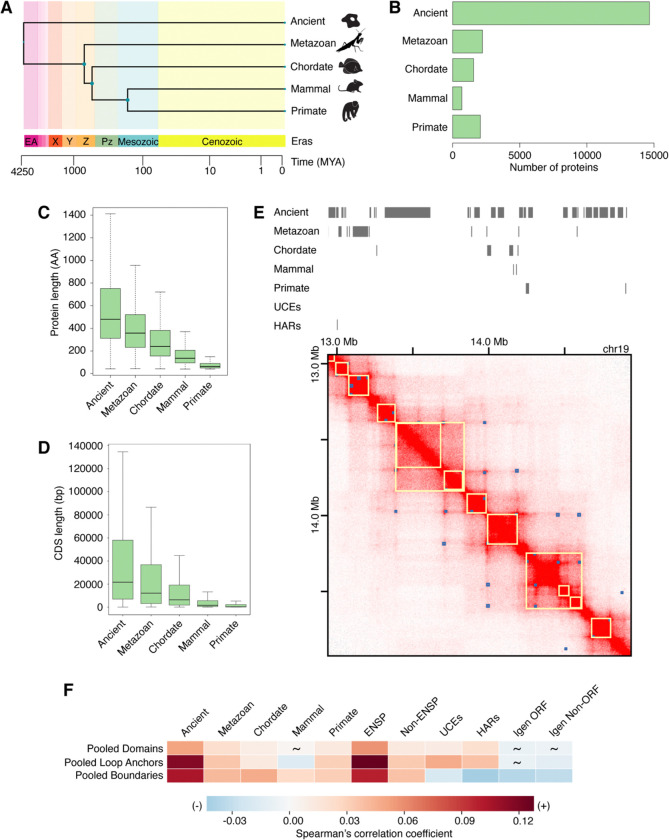
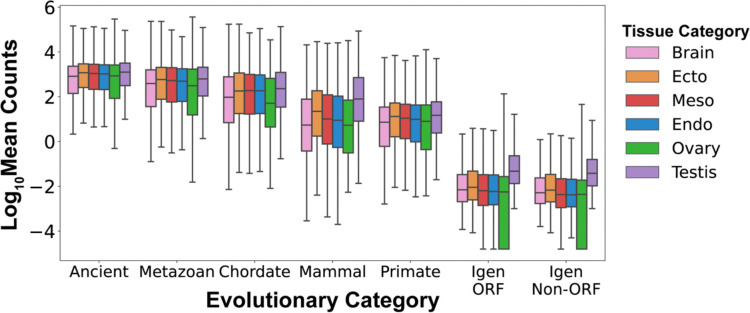
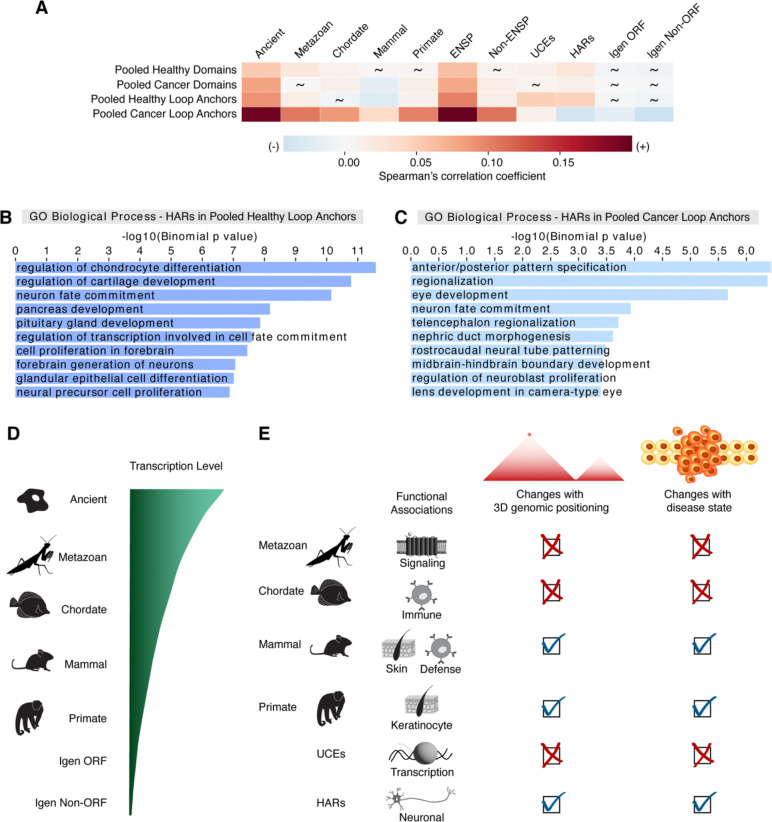
Genome organization is intricately tied to regulating genes and associated cell fate decisions. Here, we examine the positioning and functional significance of human genes, grouped by their lineage restriction level, within the 3D organization of the genome. We reveal that genes of different lineage restriction levels have distinct positioning relationships with both domains and loop anchors, and remarkably consistent relationships with boundaries across cell types. While the functional associations of each group of genes are primarily cell type-specific, associations of conserved genes maintain greater stability across 3D genomic features and disease than recently evolved genes. Furthermore, the expression of these genes across various tissues follows an evolutionary progression, such that RNA levels increase from young lineage restricted genes to ancient genes present in most species. Thus, the distinct relationships of gene evolutionary age, function, and positioning within 3D genomic features contribute to tissue-specific gene regulation in development and disease.
Keywords: 3D genome organization; cell type-specific processes; gene evolution; gene function/ontology; human accelerated regions (HARs).
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures

Similar articles
-
Topologically associating domain boundaries that are stable across diverse cell types are evolutionarily constrained and enriched for heritability.Am J Hum Genet. 2021 Feb 4;108(2):269-283. doi: 10.1016/j.ajhg.2021.01.001. Am J Hum Genet. 2021. PMID: 33545030 Free PMC article.
-
Molecular diversity and phenotypic pleiotropy of ancient genomic regulatory loci derived from human endogenous retrovirus type H (HERVH) promoter LTR7 and HERVK promoter LTR5_Hs and their contemporary impacts on pathophysiology of Modern Humans.Mol Genet Genomics. 2022 Nov;297(6):1711-1740. doi: 10.1007/s00438-022-01954-7. Epub 2022 Sep 19. Mol Genet Genomics. 2022. PMID: 36121513 Free PMC article.
-
Comparative three-dimensional genome architectures of adipose tissues provide insight into human-specific regulation of metabolic homeostasis.J Biol Chem. 2023 Jun;299(6):104757. doi: 10.1016/j.jbc.2023.104757. Epub 2023 Apr 27. J Biol Chem. 2023. PMID: 37116707 Free PMC article.
-
The role of 3D chromatin domains in gene regulation: a multi-facetted view on genome organization.Curr Opin Genet Dev. 2020 Apr;61:1-8. doi: 10.1016/j.gde.2020.02.015. Epub 2020 Mar 19. Curr Opin Genet Dev. 2020. PMID: 32199341 Review.
-
The 3D Genome in Brain Development: An Exploration of Molecular Mechanisms and Experimental Methods.Neurosci Insights. 2024 Oct 29;19:26331055241293455. doi: 10.1177/26331055241293455. eCollection 2024. Neurosci Insights. 2024. PMID: 39494115 Free PMC article. Review.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources