

This is a preprint.
Cell Marker Accordion: interpretable single-cell and spatial omics annotation in health and disease
- PMID: 38559181
- PMCID: PMC10979856
- DOI: 10.1101/2024.03.08.584053
Cell Marker Accordion: interpretable single-cell and spatial omics annotation in health and disease
Update in
-
Cell Marker Accordion: interpretable single-cell and spatial omics annotation in health and disease.Nat Commun. 2025 Jul 7;16(1):5399. doi: 10.1038/s41467-025-60900-4. Nat Commun. 2025. PMID: 40623970 Free PMC article.
Abstract
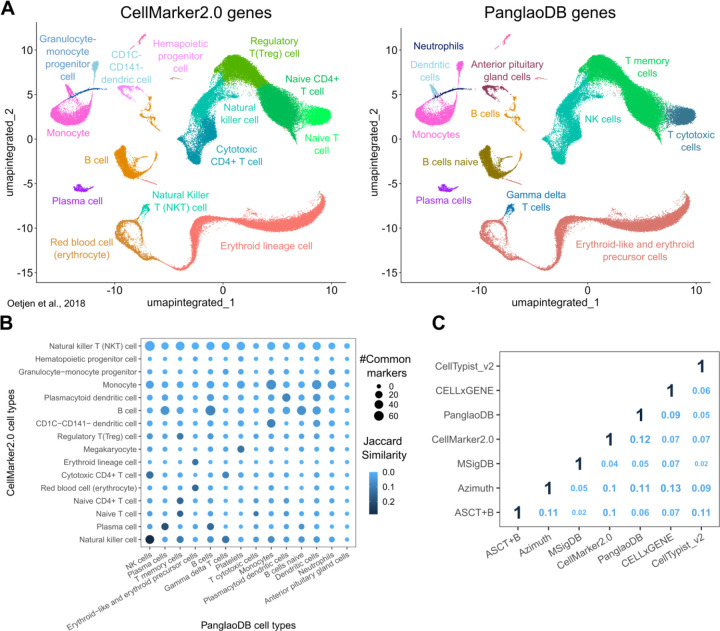
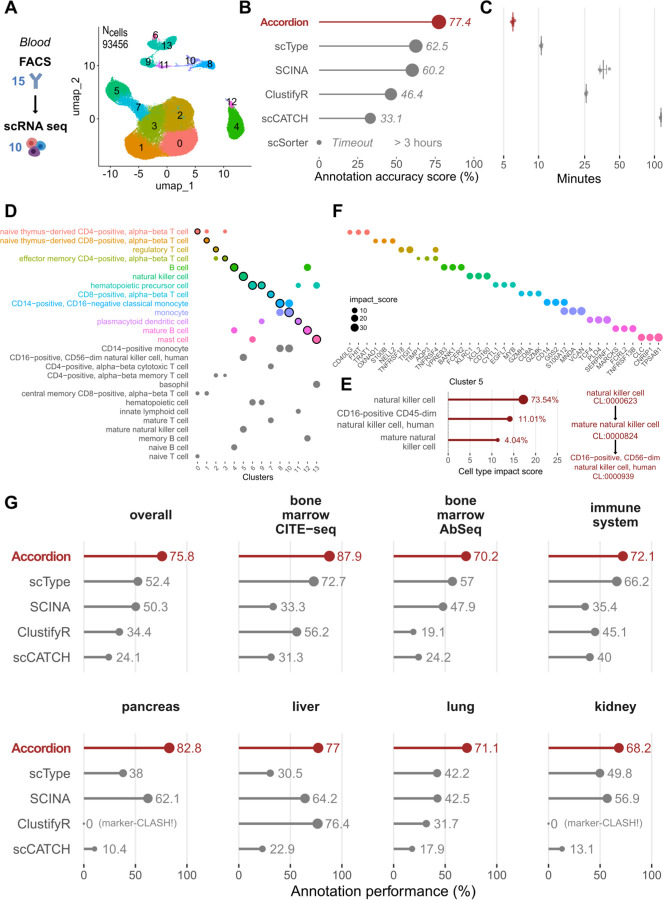
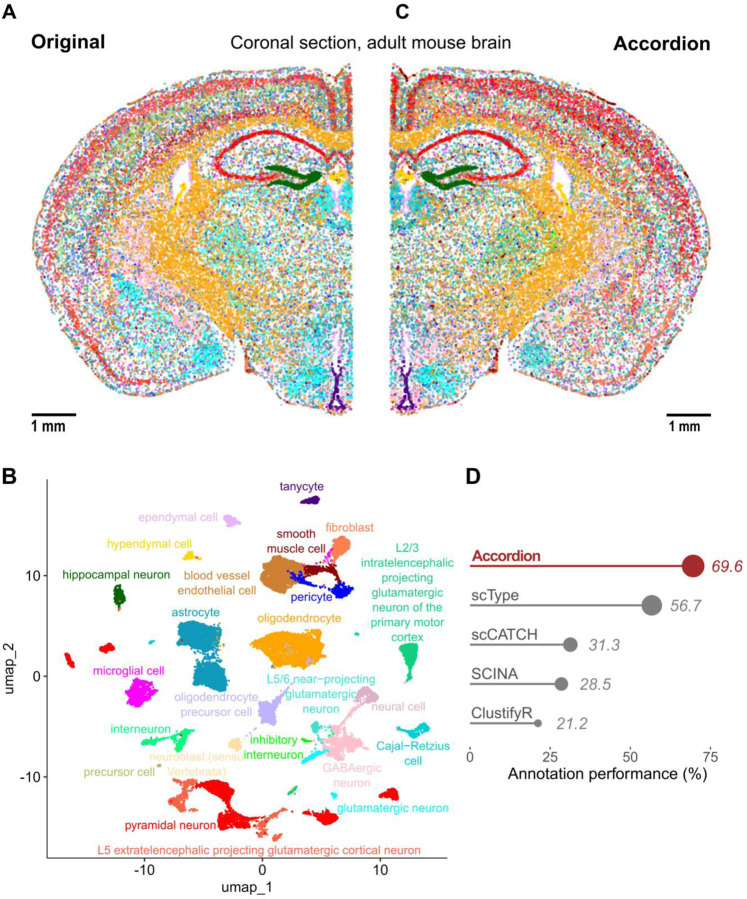
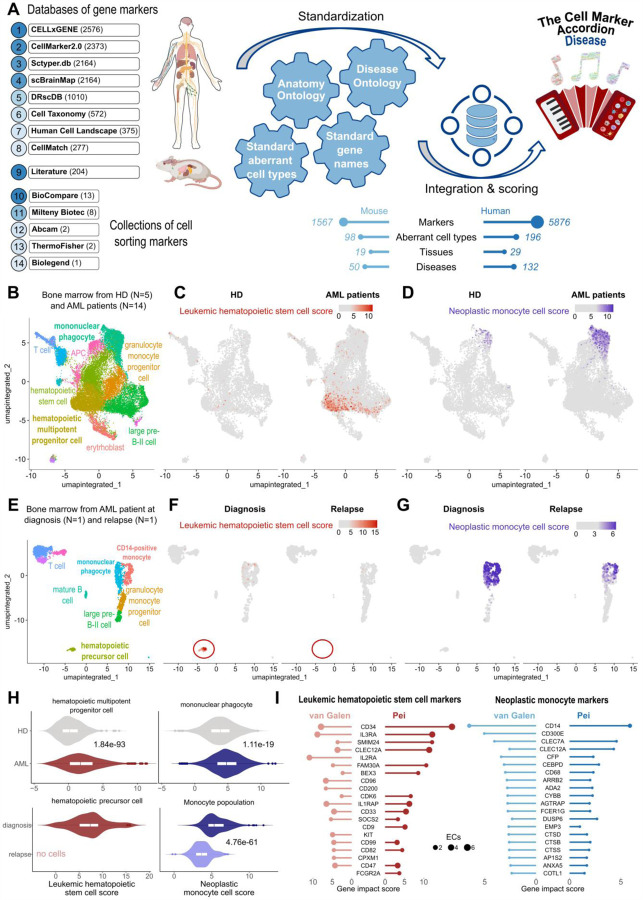
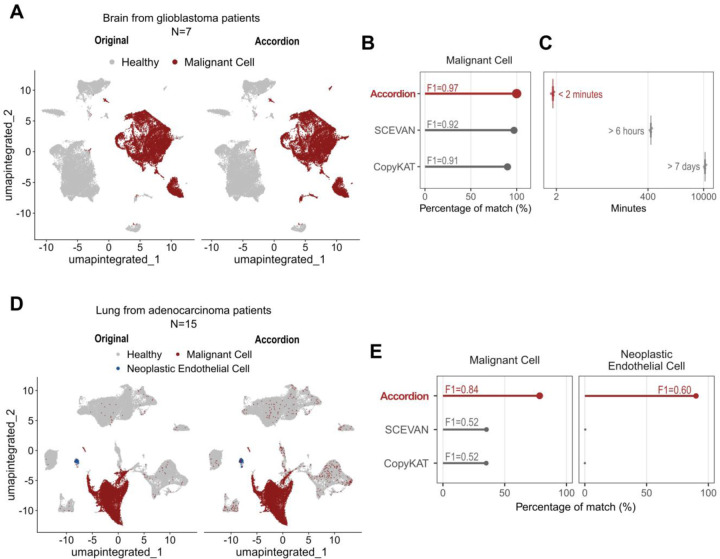
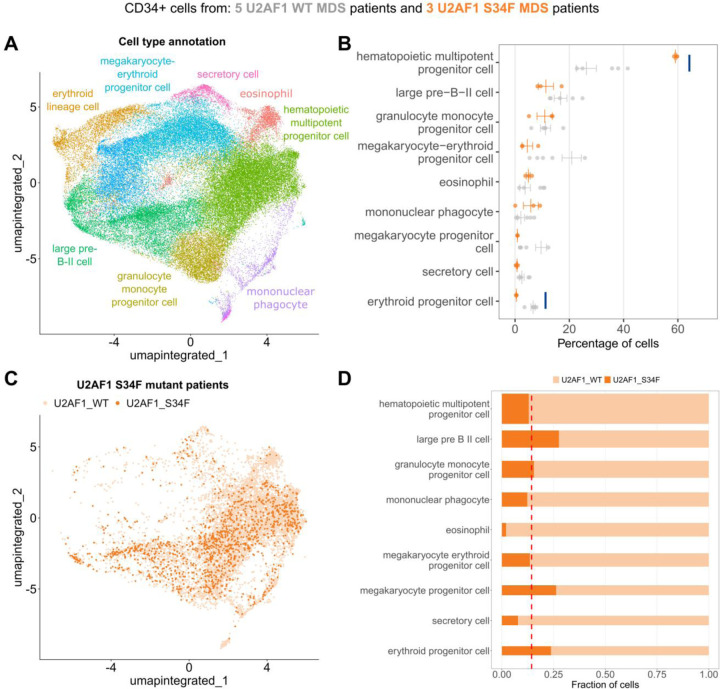
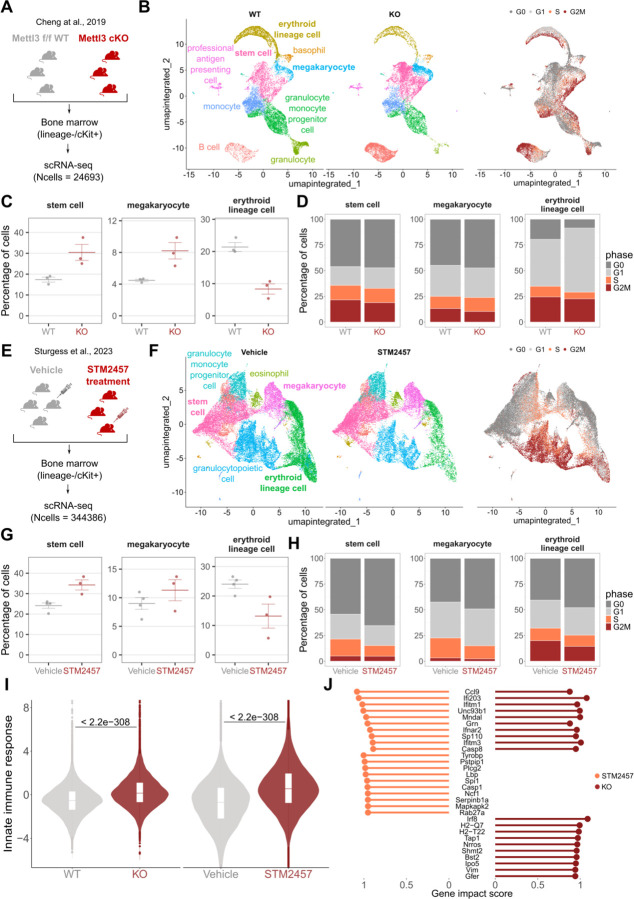
Single-cell technologies offer a unique opportunity to explore cellular heterogeneity in health and disease. However, reliable identification of cell types and states represents a bottleneck. Available databases and analysis tools employ dissimilar markers, leading to inconsistent annotations and poor interpretability. Furthermore, current tools focus mostly on physiological cell types, limiting their applicability to disease. We developed the Cell Marker Accordion, a user-friendly platform providing automatic annotation and unmatched biological interpretation of single-cell populations, based on consistency weighted markers. We validated our approach on multiple single-cell and spatial datasets from different human and murine tissues, improving annotation accuracy in all cases. Moreover, we show that the Cell Marker Accordion can identify disease-critical cells and pathological processes, extracting potential biomarkers in a wide variety of disease contexts. The breadth of these applications elevates the Cell Marker Accordion as a fast, flexible, faithful and standardized tool to annotate and interpret single-cell and spatial populations in studying physiology and disease.
Conflict of interest statement
Competing Interests S.H., consultancy, Forma Therapeutics. Other authors declare no competing financial interests.
Figures

Similar articles
-
Cell Marker Accordion: interpretable single-cell and spatial omics annotation in health and disease.Nat Commun. 2025 Jul 7;16(1):5399. doi: 10.1038/s41467-025-60900-4. Nat Commun. 2025. PMID: 40623970 Free PMC article.
-
Leveraging a foundation model zoo for cell similarity search in oncological microscopy across devices.Front Oncol. 2025 Jun 18;15:1480384. doi: 10.3389/fonc.2025.1480384. eCollection 2025. Front Oncol. 2025. PMID: 40606969 Free PMC article.
-
Measures implemented in the school setting to contain the COVID-19 pandemic.Cochrane Database Syst Rev. 2022 Jan 17;1(1):CD015029. doi: 10.1002/14651858.CD015029. Cochrane Database Syst Rev. 2022. Update in: Cochrane Database Syst Rev. 2024 May 2;5:CD015029. doi: 10.1002/14651858.CD015029.pub2. PMID: 35037252 Free PMC article. Updated.
-
ScInfeR: an efficient method for annotating cell types and sub-types in single-cell RNA-seq, ATAC-seq, and spatial omics.Brief Bioinform. 2025 May 1;26(3):bbaf253. doi: 10.1093/bib/bbaf253. Brief Bioinform. 2025. PMID: 40471991 Free PMC article.
-
Comparison of self-administered survey questionnaire responses collected using mobile apps versus other methods.Cochrane Database Syst Rev. 2015 Jul 27;2015(7):MR000042. doi: 10.1002/14651858.MR000042.pub2. Cochrane Database Syst Rev. 2015. PMID: 26212714 Free PMC article.
References
-
- Monga I., Kaur K. & Dhanda S. K. Revisiting hematopoiesis: applications of the bulk and single-cell transcriptomics dissecting transcriptional heterogeneity in hematopoietic stem cells. Brief Funct Genomics 21, 159–176 (2022). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources