

Complexity of avian evolution revealed by family-level genomes
- PMID: 38560995
- PMCID: PMC11111414
- DOI: 10.1038/s41586-024-07323-1
Complexity of avian evolution revealed by family-level genomes
Abstract
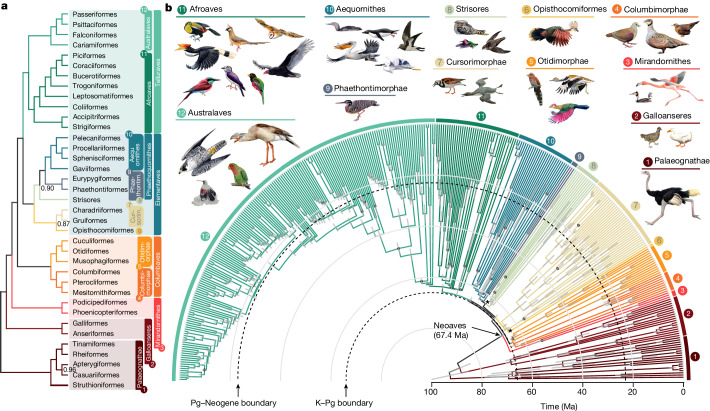
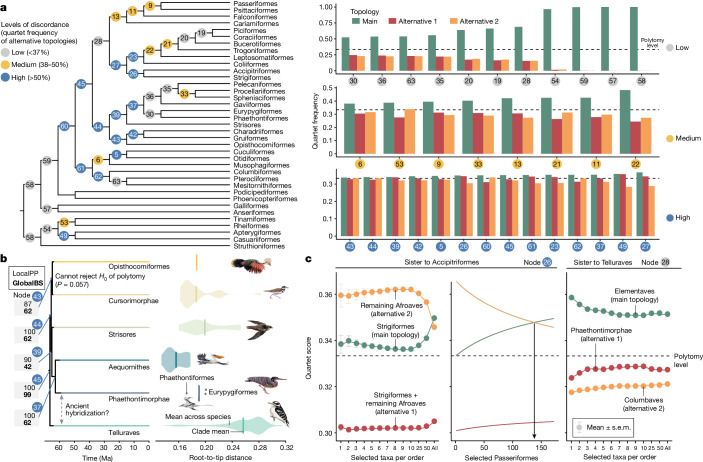
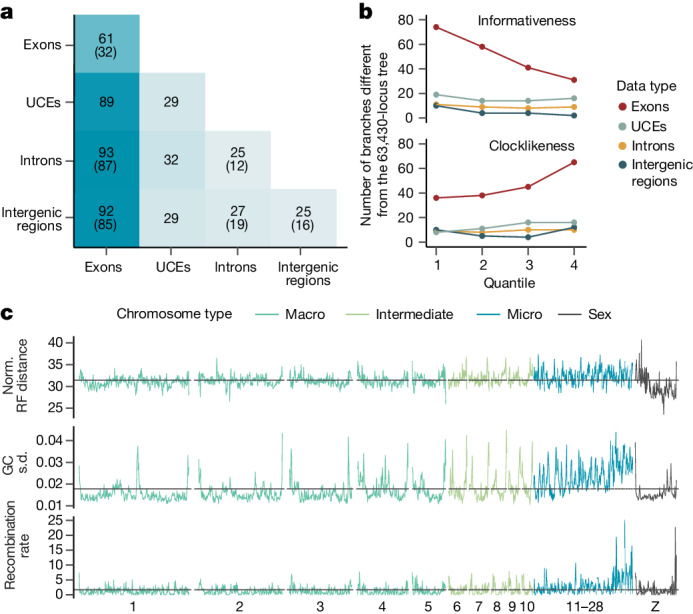
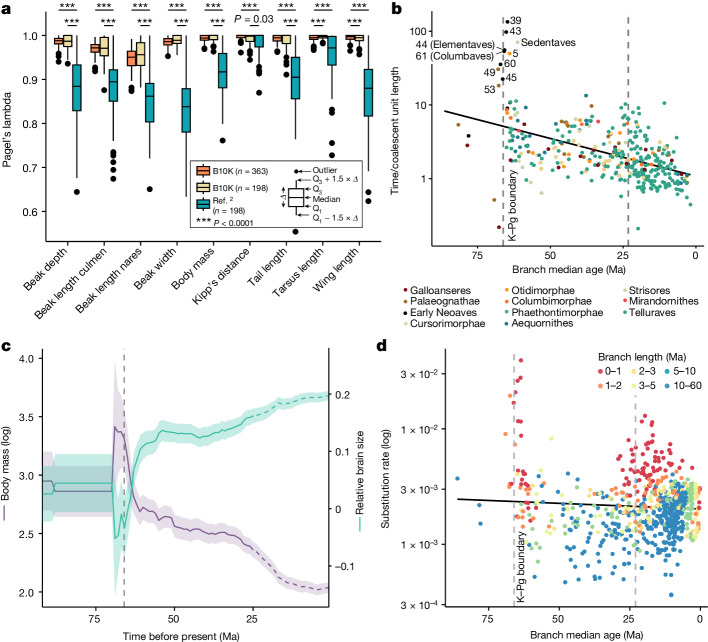
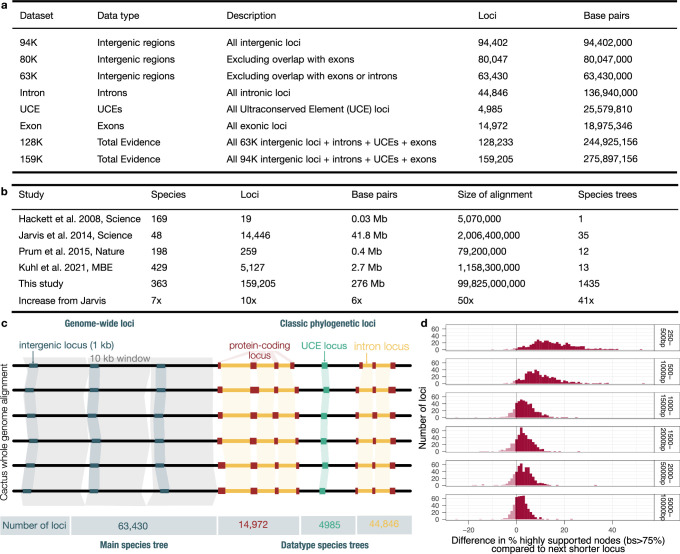
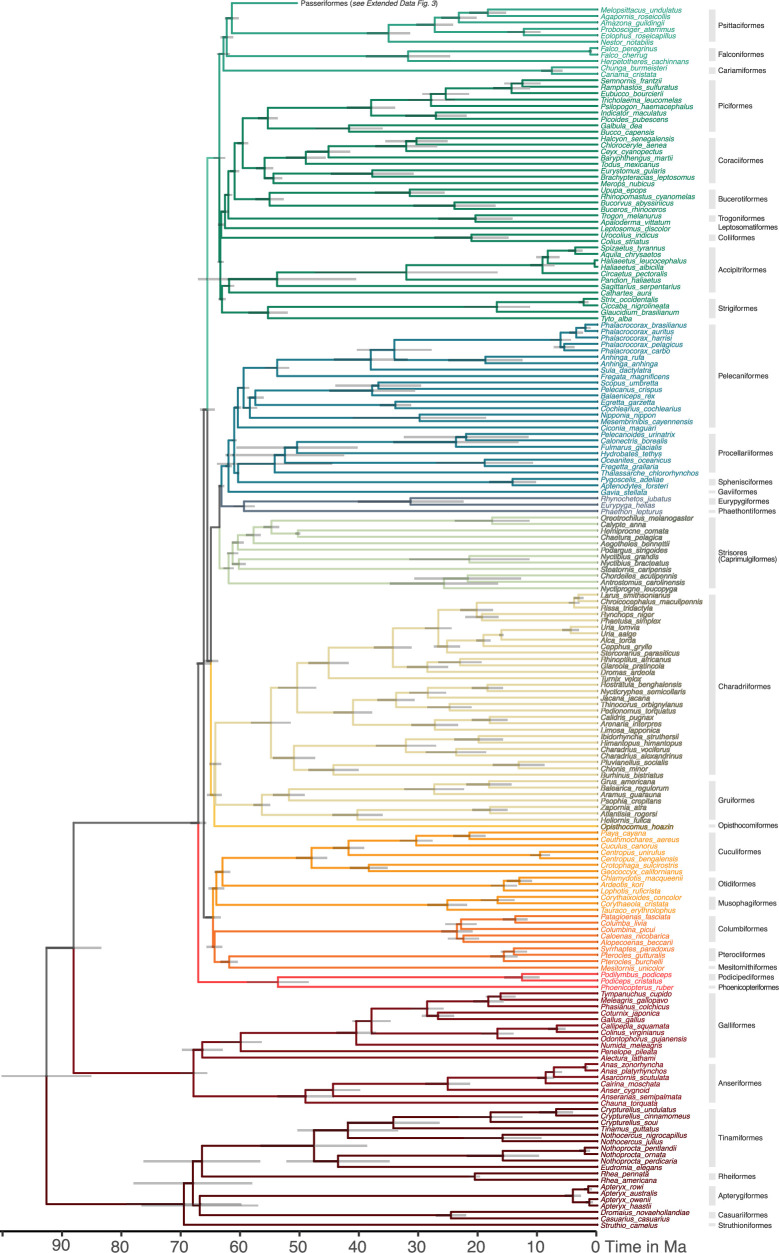
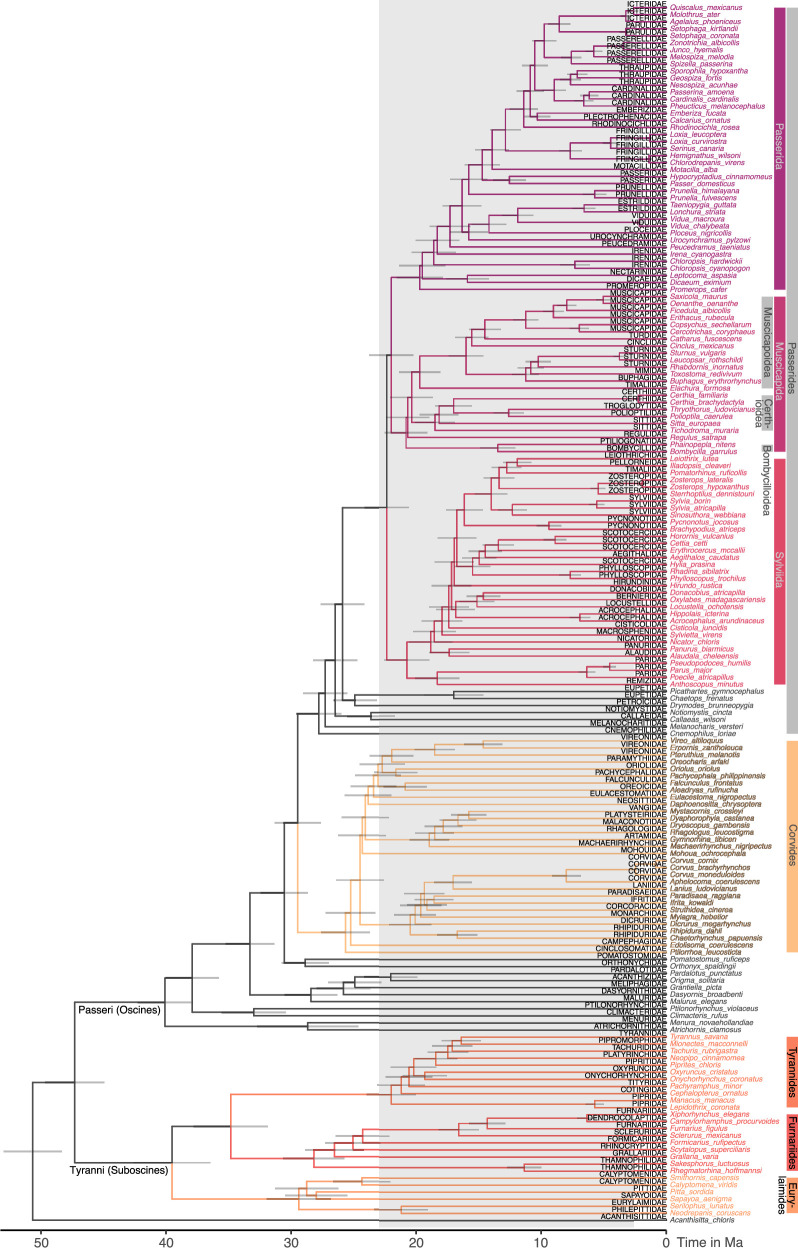
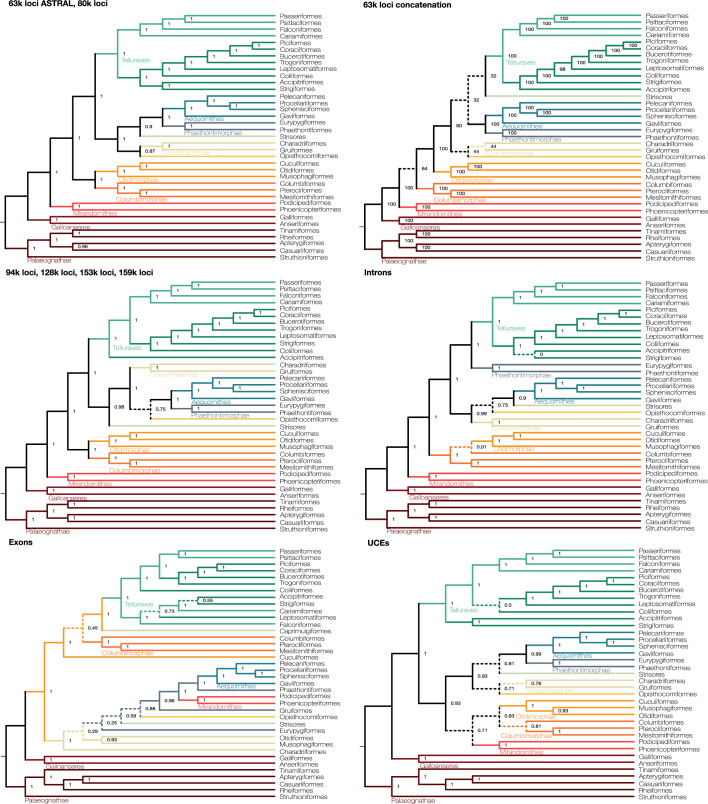
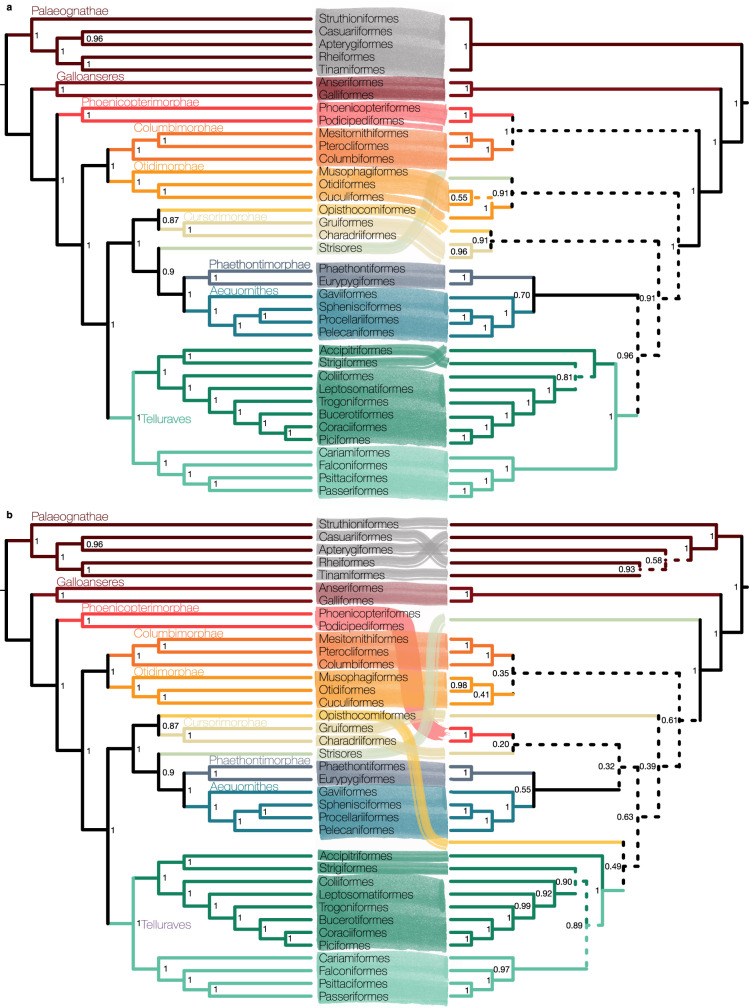
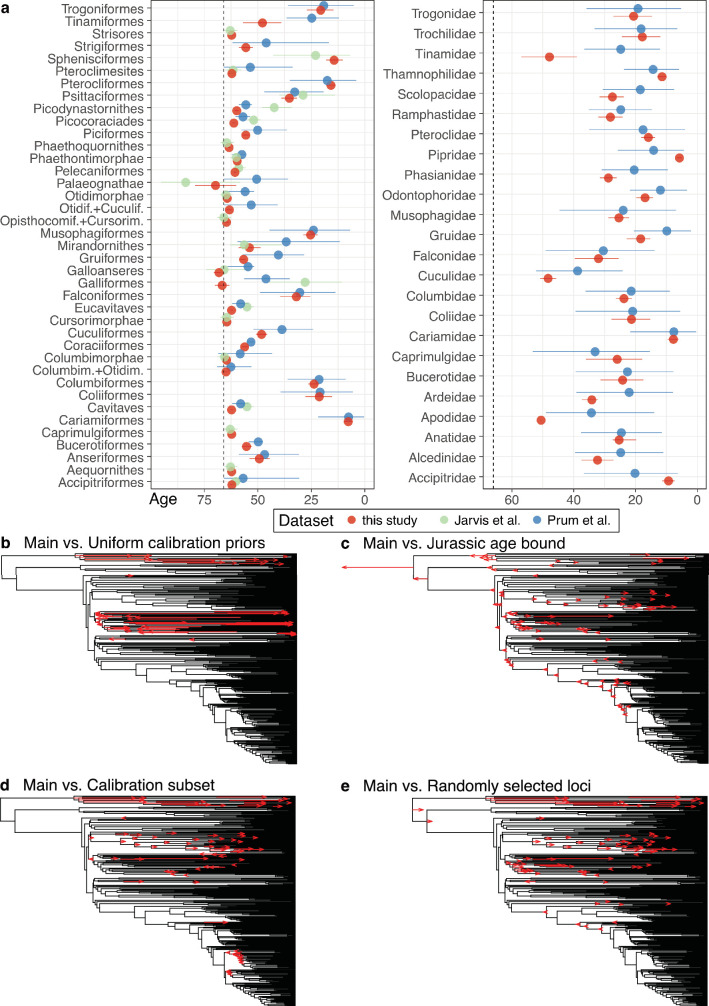
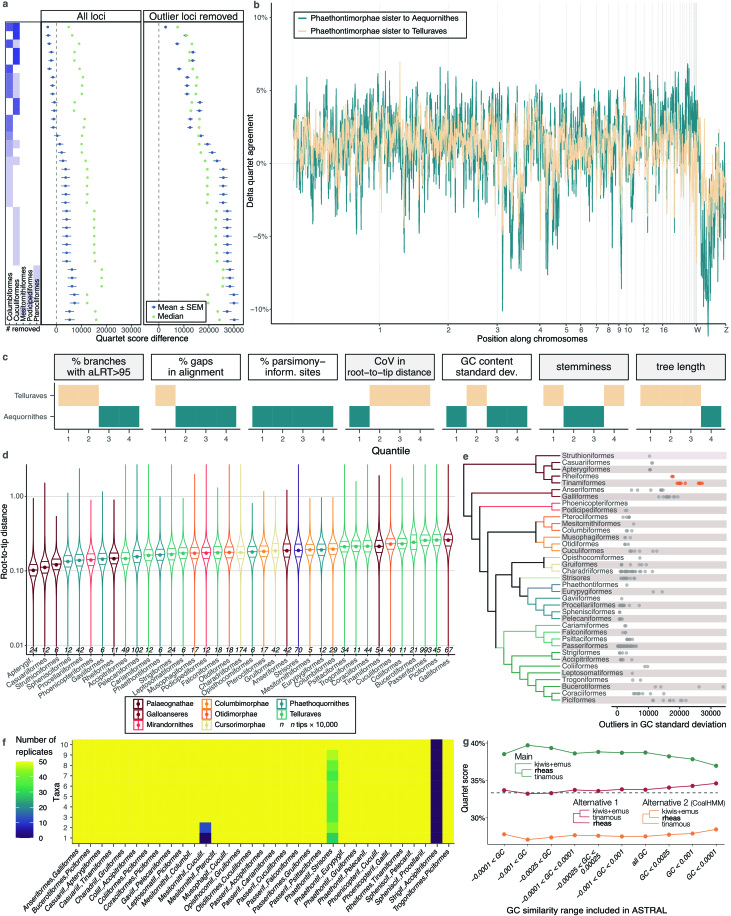
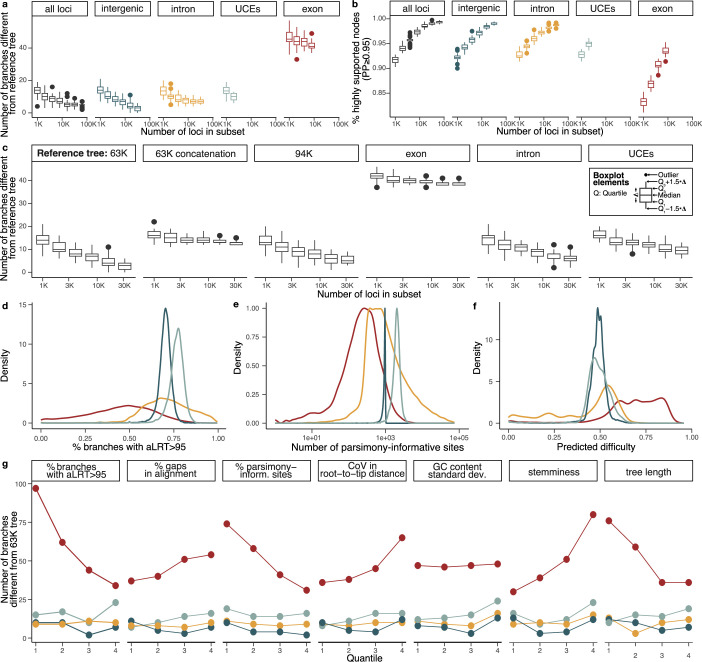
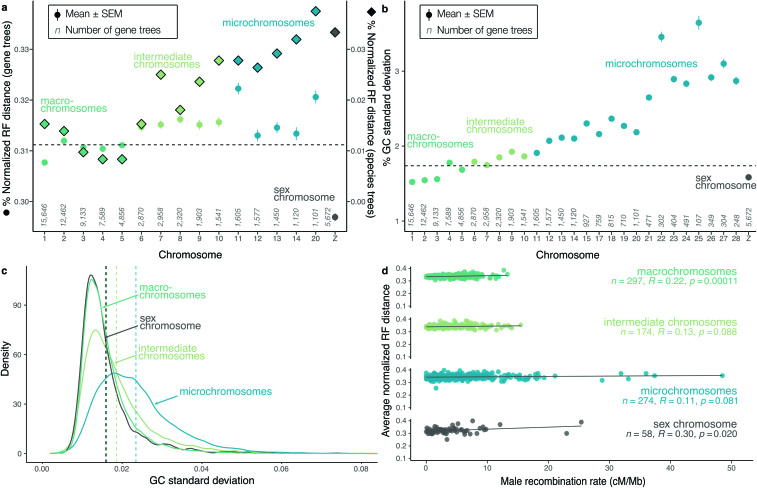
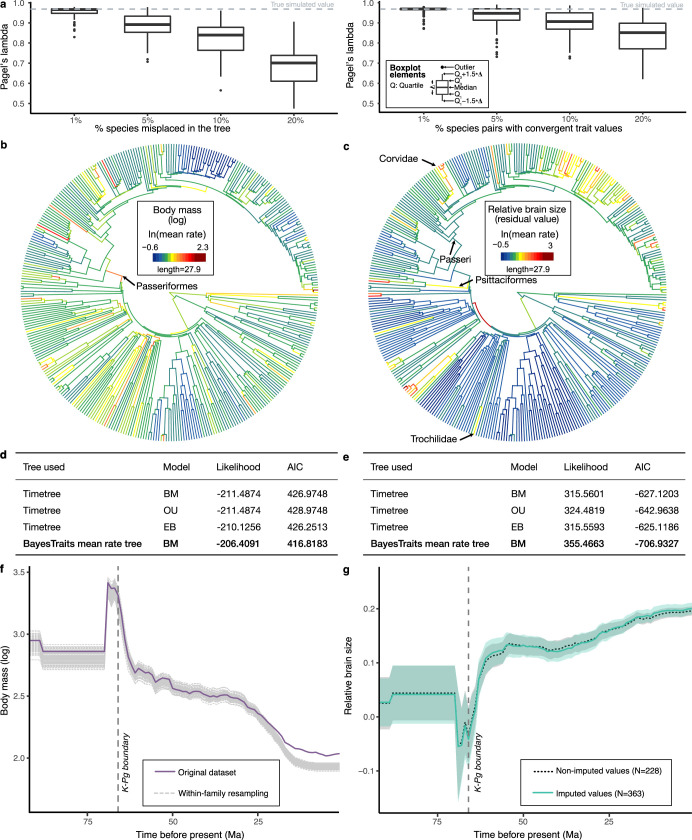
Despite tremendous efforts in the past decades, relationships among main avian lineages remain heavily debated without a clear resolution. Discrepancies have been attributed to diversity of species sampled, phylogenetic method and the choice of genomic regions1-3. Here we address these issues by analysing the genomes of 363 bird species4 (218 taxonomic families, 92% of total). Using intergenic regions and coalescent methods, we present a well-supported tree but also a marked degree of discordance. The tree confirms that Neoaves experienced rapid radiation at or near the Cretaceous-Palaeogene boundary. Sufficient loci rather than extensive taxon sampling were more effective in resolving difficult nodes. Remaining recalcitrant nodes involve species that are a challenge to model due to either extreme DNA composition, variable substitution rates, incomplete lineage sorting or complex evolutionary events such as ancient hybridization. Assessment of the effects of different genomic partitions showed high heterogeneity across the genome. We discovered sharp increases in effective population size, substitution rates and relative brain size following the Cretaceous-Palaeogene extinction event, supporting the hypothesis that emerging ecological opportunities catalysed the diversification of modern birds. The resulting phylogenetic estimate offers fresh insights into the rapid radiation of modern birds and provides a taxon-rich backbone tree for future comparative studies.
© 2024. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
M.T.P.G. serves on the Science Advisory Board of Colossal Laboratories & Biosciences. All other authors declare no competing interests.
Figures


References
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
