

Nucleosome reorganisation in breast cancer tissues
- PMID: 38561804
- PMCID: PMC10986098
- DOI: 10.1186/s13148-024-01656-4
Nucleosome reorganisation in breast cancer tissues
Abstract
Background: Nucleosome repositioning in cancer is believed to cause many changes in genome organisation and gene expression. Understanding these changes is important to elucidate fundamental aspects of cancer. It is also important for medical diagnostics based on cell-free DNA (cfDNA), which originates from genomic DNA regions protected from digestion by nucleosomes.
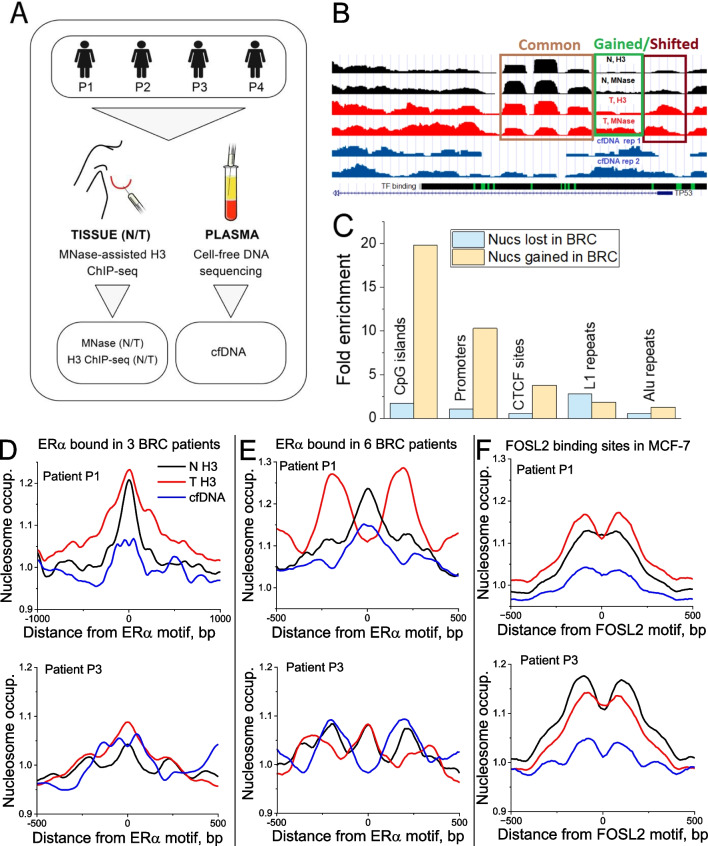
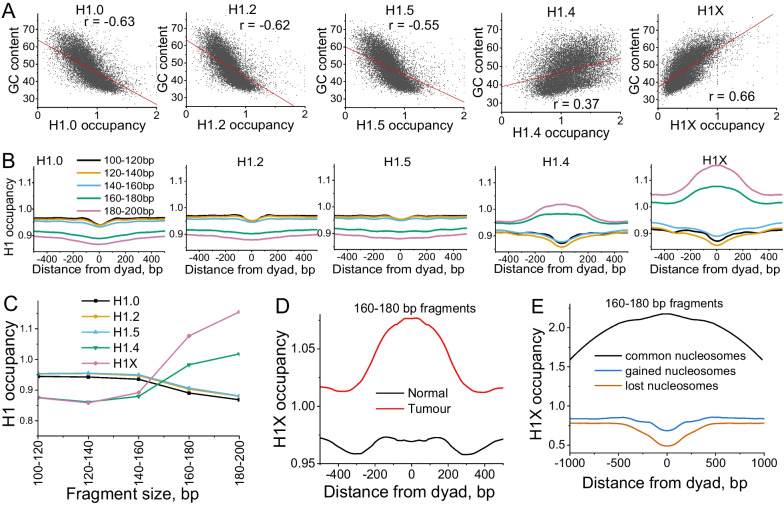
Results: We have generated high-resolution nucleosome maps in paired tumour and normal tissues from the same breast cancer patients using MNase-assisted histone H3 ChIP-seq and compared them with the corresponding cfDNA from blood plasma. This analysis has detected single-nucleosome repositioning at key regulatory regions in a patient-specific manner and common cancer-specific patterns across patients. The nucleosomes gained in tumour versus normal tissue were particularly informative of cancer pathways, with ~ 20-fold enrichment at CpG islands, a large fraction of which marked promoters of genes encoding DNA-binding proteins. The tumour tissues were characterised by a 5-10 bp decrease in the average distance between nucleosomes (nucleosome repeat length, NRL), which is qualitatively similar to the differences between pluripotent and differentiated cells. This effect was correlated with gene activity, differential DNA methylation and changes in local occupancy of linker histone variants H1.4 and H1X.
Conclusions: Our study offers a novel resource of high-resolution nucleosome maps in breast cancer patients and reports for the first time the effect of systematic decrease of NRL in paired tumour versus normal breast tissues from the same patient. Our findings provide a new mechanistic understanding of nucleosome repositioning in tumour tissues that can be valuable for patient diagnostics, stratification and monitoring.
Keywords: Breast cancer; Chromatin; Linker histones; Liquid biopsy; NRL; Nucleosome positioning; Nucleosome repeat length; Nucleosomics; Transcription factors binding; cfDNA.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
