

This is a preprint.
Atomically accurate de novo design of antibodies with RFdiffusion
- PMID: 38562682
- PMCID: PMC10983868
- DOI: 10.1101/2024.03.14.585103
Atomically accurate de novo design of antibodies with RFdiffusion
Update in
-
Atomically accurate de novo design of antibodies with RFdiffusion.Nature. 2025 Nov 5. doi: 10.1038/s41586-025-09721-5. Online ahead of print. Nature. 2025. PMID: 41193805
Abstract
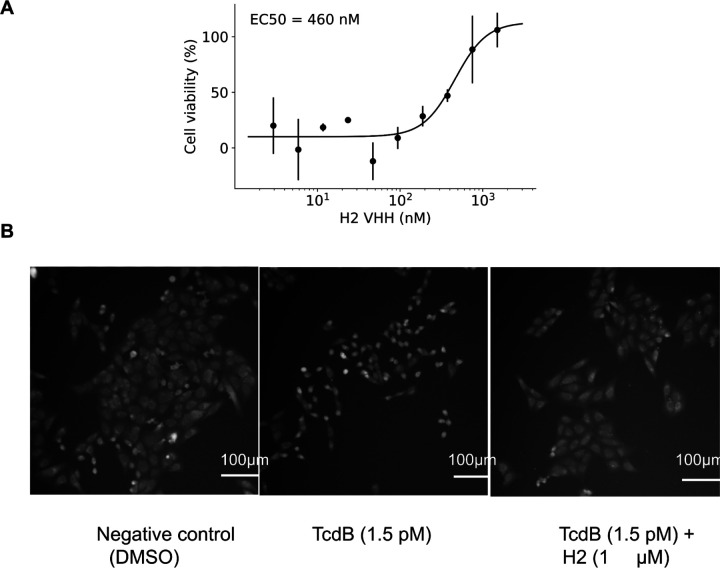
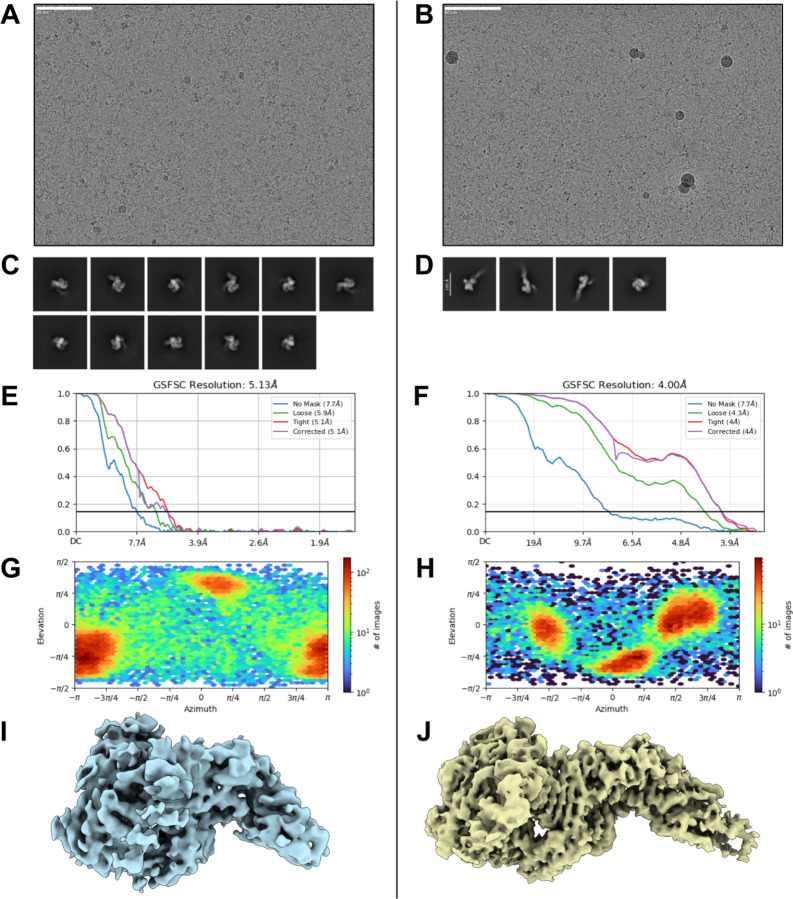
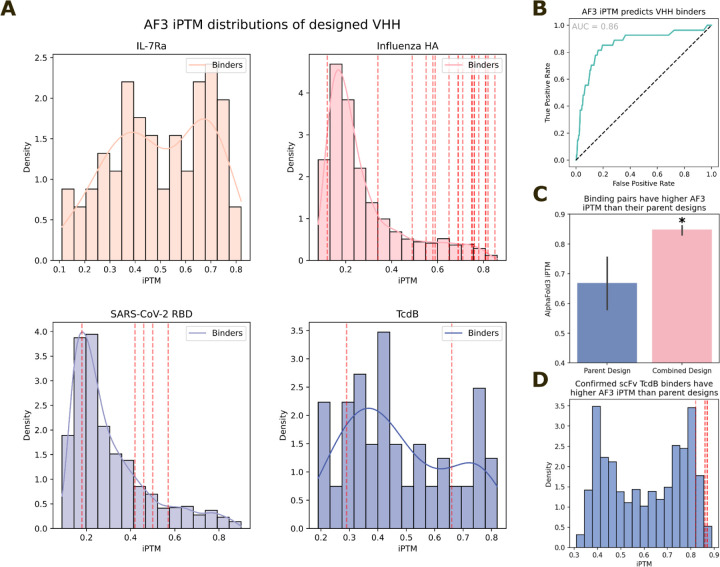
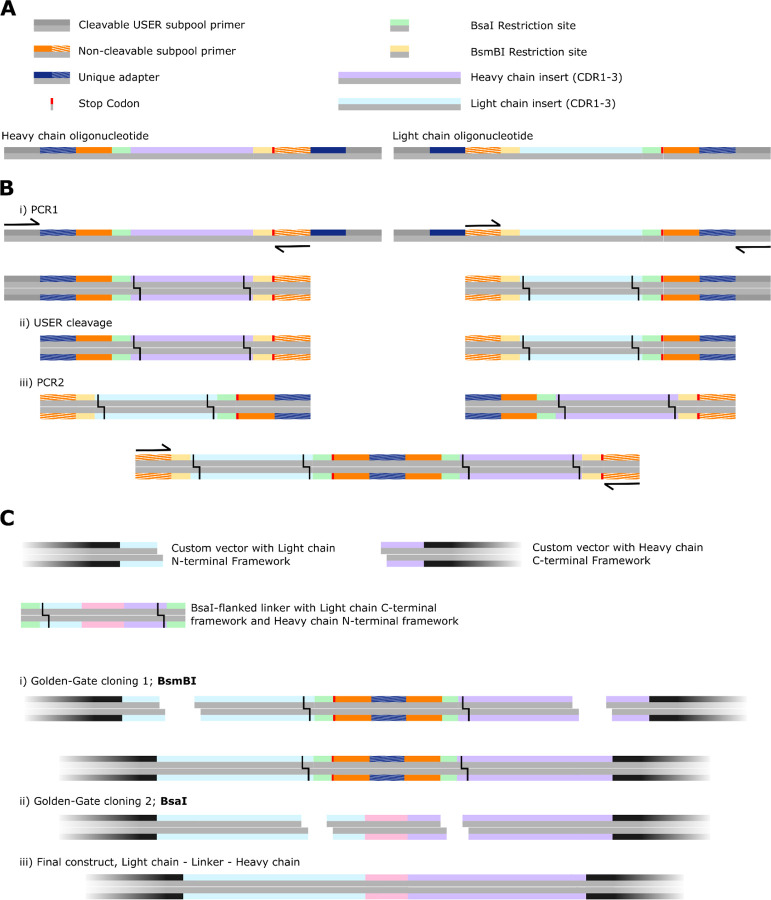
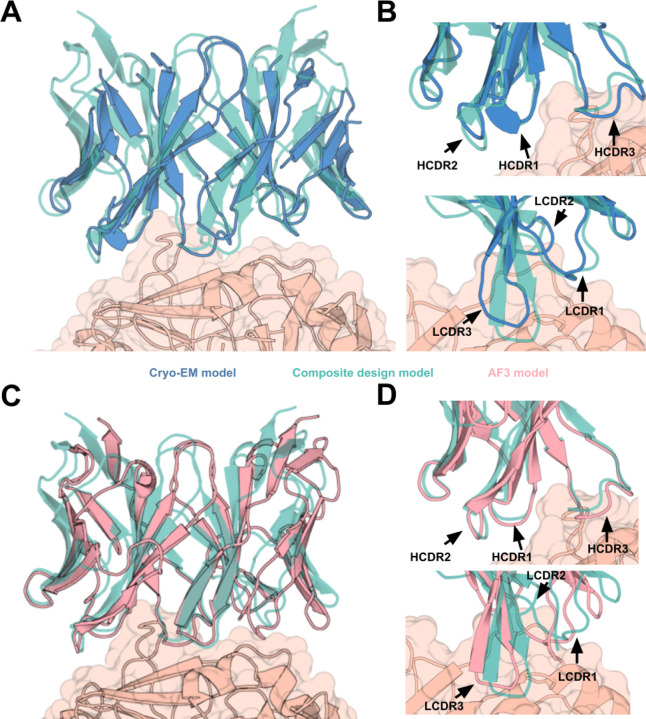
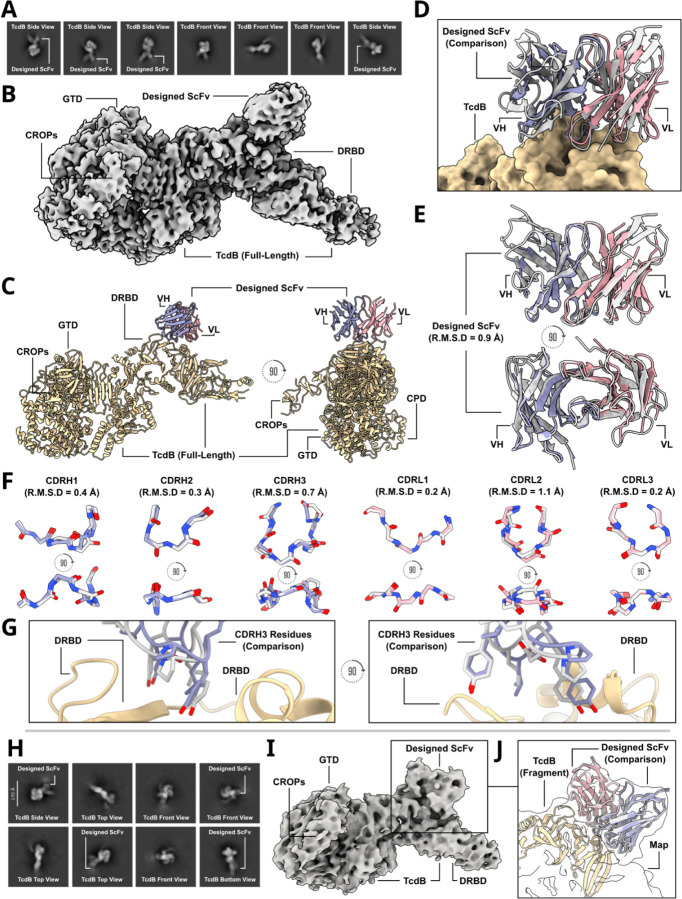
Despite the central role that antibodies play in modern medicine, there is currently no method to design novel antibodies that bind a specific epitope entirely in silico. Instead, antibody discovery currently relies on animal immunization or random library screening approaches. Here, we demonstrate that combining computational protein design using a fine-tuned RFdiffusion network alongside yeast display screening enables the generation of antibody variable heavy chains (VHHs) and single chain variable fragments (scFvs) that bind user-specified epitopes with atomic-level precision. To verify this, we experimentally characterized VHH binders to four disease-relevant epitopes using multiple orthogonal biophysical methods, including cryo-EM, which confirmed the proper Ig fold and binding pose of designed VHHs targeting influenza hemagglutinin and Clostridium difficile toxin B (TcdB). For the influenza-targeting VHH, high-resolution structural data further confirmed the accuracy of CDR loop conformations. While initial computational designs exhibit modest affinity, affinity maturation using OrthoRep enables production of single-digit nanomolar binders that maintain the intended epitope selectivity. We further demonstrate the de novo design of single-chain variable fragments (scFvs), creating binders to TcdB and a Phox2b peptide-MHC complex by combining designed heavy and light chain CDRs. Cryo-EM structural data confirmed the proper Ig fold and binding pose for two distinct TcdB scFvs, with high-resolution data for one design additionally verifying the atomically accurate conformations of all six CDR loops. Our approach establishes a framework for the rational computational design, screening, isolation, and characterization of fully de novo antibodies with atomic-level precision in both structure and epitope targeting.
Conflict of interest statement
Competing Interests N.R.B., J.L.W., R.J.R., A.J.B., C.W., P.J.Y.L., B.H., and D.B. are co-inventors on U.S. provisional patent number 63/607,651 which covers the computational antibody design pipeline described here. N.R.B., J.L.W, P.J.Y.L. and B.H. are currently employed by Xaira Therapeutics. N.R.B., J.L.W, P.J.Y.L., B.H., R.J.R, A.J.B., and C.W. have received payments relating to the licensing of the inventions described here to Xaira Therapeutics. C.C.L. is a co-founder of K2 Therapeutics, which uses OrthoRep in antibody engineering and evolution.
Figures




























References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials