

This is a preprint.
Transcriptional Determinism and Stochasticity Contribute to the Complexity of Autism Associated SHANK Family Genes
- PMID: 38562714
- PMCID: PMC10983920
- DOI: 10.1101/2024.03.18.585480
Transcriptional Determinism and Stochasticity Contribute to the Complexity of Autism Associated SHANK Family Genes
Update in
-
Transcriptional determinism and stochasticity contribute to the complexity of autism-associated SHANK family genes.Cell Rep. 2024 Jul 23;43(7):114376. doi: 10.1016/j.celrep.2024.114376. Epub 2024 Jun 18. Cell Rep. 2024. PMID: 38900637 Free PMC article.
Abstract
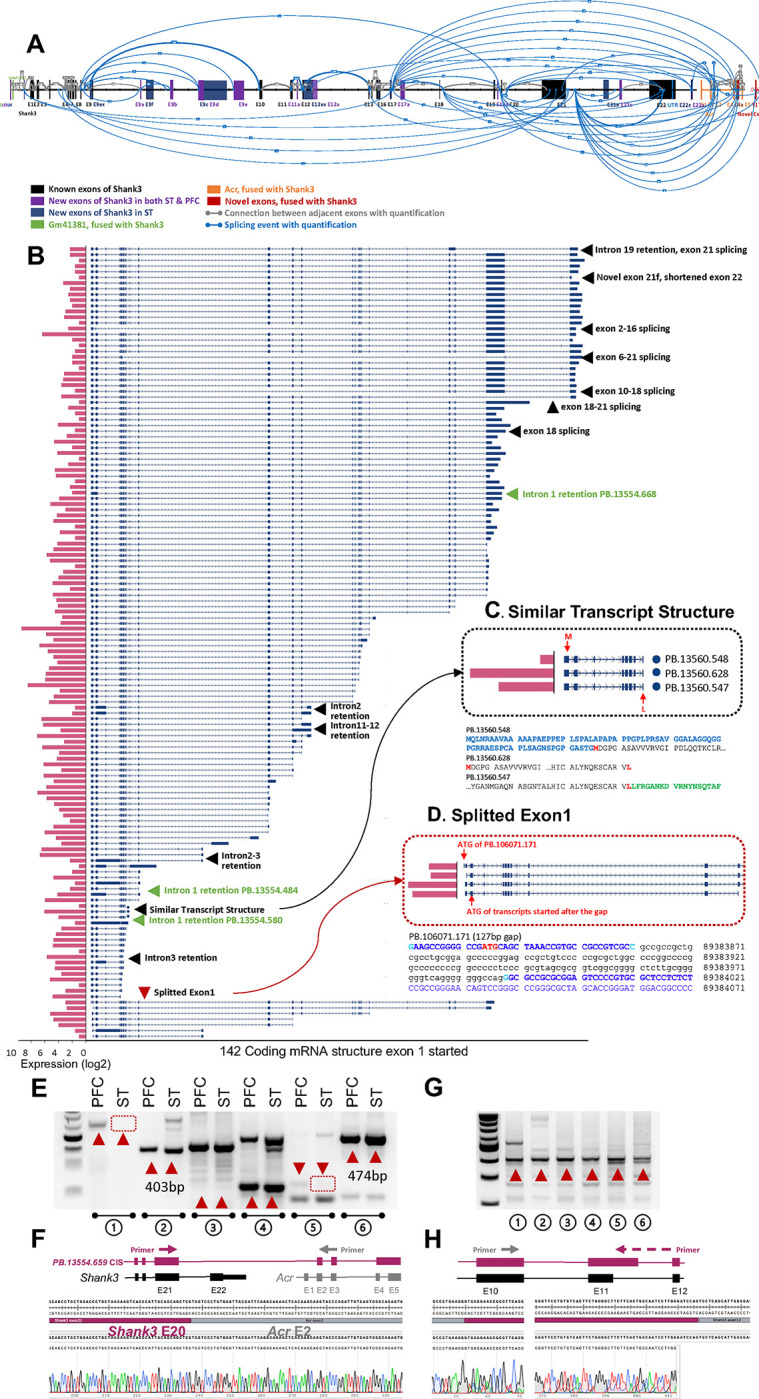
Precision of transcription is critical because transcriptional dysregulation is disease causing. Traditional methods of transcriptional profiling are inadequate to elucidate the full spectrum of the transcriptome, particularly for longer and less abundant mRNAs. SHANK3 is one of the most common autism causative genes. Twenty-four Shank3 mutant animal lines have been developed for autism modeling. However, their preclinical validity has been questioned due to incomplete Shank3 transcript structure. We applied an integrative approach combining cDNA-capture and long-read sequencing to profile the SHANK3 transcriptome in human and mice. We unexpectedly discovered an extremely complex SHANK3 transcriptome. Specific SHANK3 transcripts were altered in Shank3 mutant mice and postmortem brains tissues from individuals with ASD. The enhanced SHANK3 transcriptome significantly improved the detection rate for potential deleterious variants from genomics studies of neuropsychiatric disorders. Our findings suggest the stochastic transcription of genome associated with SHANK family genes.
Conflict of interest statement
Competing interests YHJ is a scientific co-founder of Couragene. Inc but this study is unrelated to his role. The project was supported initially by sponsored research project by Taysha Gene Therapies. Taysha Gene Therapies did not have any direct tole for the conceptualization, design, data collection, analysis, decision to publish, or preparation of the manuscript.
Figures






References
-
- Ray T.A., Cochran K., Kozlowski C., Wang J., Alexander G., Cady M.A., Spencer W.J., Ruzycki P.A., Clark B.S., Laeremans A., et al. (2020). Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat Commun 11, 3328. 10.1038/s41467-020-17009-7. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources