

This is a preprint.
Canopy2: tumor phylogeny inference by bulk DNA and single-cell RNA sequencing
- PMID: 38562795
- PMCID: PMC10983938
- DOI: 10.1101/2024.03.18.585595
Canopy2: tumor phylogeny inference by bulk DNA and single-cell RNA sequencing
Abstract
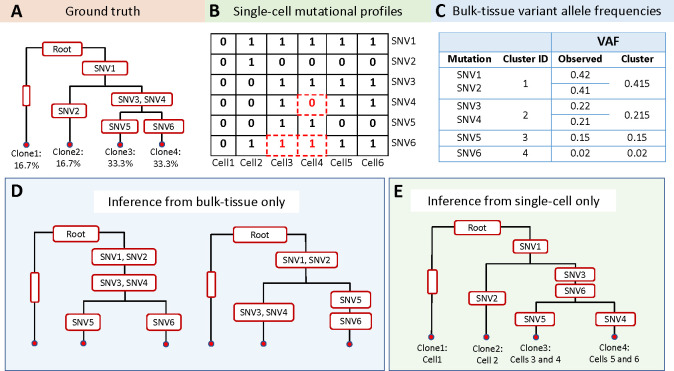
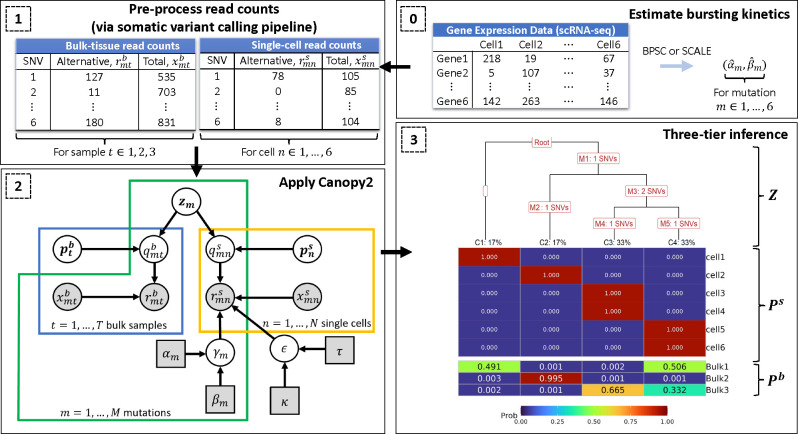
Tumors are comprised of a mixture of distinct cell populations that differ in terms of genetic makeup and function. Such heterogeneity plays a role in the development of drug resistance and the ineffectiveness of targeted cancer therapies. Insight into this complexity can be obtained through the construction of a phylogenetic tree, which illustrates the evolutionary lineage of tumor cells as they acquire mutations over time. We propose Canopy2, a Bayesian framework that uses single nucleotide variants derived from bulk DNA and single-cell RNA sequencing to infer tumor phylogeny and conduct mutational profiling of tumor subpopulations. Canopy2 uses Markov chain Monte Carlo methods to sample from a joint probability distribution involving a mixture of binomial and beta-binomial distributions, specifically chosen to account for the sparsity and stochasticity of the single-cell data. Canopy2 demystifies the sources of zeros in the single-cell data and separates zeros categorized as non-cancerous (cells without mutations), stochastic (mutations not expressed due to bursting), and technical (expressed mutations not picked up by sequencing). Simulations demonstrate that Canopy2 consistently outperforms competing methods and reconstructs the clonal tree with high fidelity, even in situations involving low sequencing depth, poor single-cell yield, and highly-advanced and polyclonal tumors. We further assess the performance of Canopy2 through application to breast cancer and glioblastoma data, benchmarking against existing methods. Canopy2 is an open-source R package available at https://github.com/annweideman/canopy2.
Figures



Similar articles
-
BiTSC 2: Bayesian inference of tumor clonal tree by joint analysis of single-cell SNV and CNA data.Brief Bioinform. 2022 May 13;23(3):bbac092. doi: 10.1093/bib/bbac092. Brief Bioinform. 2022. PMID: 35368055 Free PMC article.
-
Conifer: clonal tree inference for tumor heterogeneity with single-cell and bulk sequencing data.BMC Bioinformatics. 2021 Aug 30;22(1):416. doi: 10.1186/s12859-021-04338-7. BMC Bioinformatics. 2021. PMID: 34461827 Free PMC article.
-
SiCloneFit: Bayesian inference of population structure, genotype, and phylogeny of tumor clones from single-cell genome sequencing data.Genome Res. 2019 Nov;29(11):1847-1859. doi: 10.1101/gr.243121.118. Epub 2019 Oct 18. Genome Res. 2019. PMID: 31628257 Free PMC article.
-
ConDoR: Tumor phylogeny inference with a copy-number constrained mutation loss model.bioRxiv [Preprint]. 2023 Jan 6:2023.01.05.522408. doi: 10.1101/2023.01.05.522408. bioRxiv. 2023. Update in: Genome Biol. 2023 Nov 30;24(1):272. doi: 10.1186/s13059-023-03106-5. PMID: 36711528 Free PMC article. Updated. Preprint.
-
Inference of clonal selection in cancer populations using single-cell sequencing data.Bioinformatics. 2019 Jul 15;35(14):i398-i407. doi: 10.1093/bioinformatics/btz392. Bioinformatics. 2019. PMID: 31510696 Free PMC article.
References
-
- Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C, Lichtenstein L. Calling somatic SNVs and indels with Mutect2. BioRxiv. 2019; p. 861054.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources