

IL2 Targeted to CD8+ T Cells Promotes Robust Effector T-cell Responses and Potent Antitumor Immunity
- PMID: 38563906
- PMCID: PMC11215410
- DOI: 10.1158/2159-8290.CD-23-1266
IL2 Targeted to CD8+ T Cells Promotes Robust Effector T-cell Responses and Potent Antitumor Immunity
Abstract
IL2 signals pleiotropically on diverse cell types, some of which contribute to therapeutic activity against tumors, whereas others drive undesired activity, such as immunosuppression or toxicity. We explored the theory that targeting of IL2 to CD8+ T cells, which are key antitumor effectors, could enhance its therapeutic index. To this aim, we developed AB248, a CD8 cis-targeted IL2 that demonstrates over 500-fold preference for CD8+ T cells over natural killer and regulatory T cells (Tregs), which may contribute to toxicity and immunosuppression, respectively. AB248 recapitulated IL2's effects on CD8+ T cells in vitro and induced selective expansion of CD8+T cells in primates. In mice, an AB248 surrogate demonstrated superior antitumor activity and enhanced tolerability as compared with an untargeted IL2Rβγ agonist. Efficacy was associated with the expansion and phenotypic enhancement of tumor-infiltrating CD8+ T cells, including the emergence of a "better effector" population. These data support the potential utility of AB248 in clinical settings. Significance: The full potential of IL2 therapy remains to be unlocked. We demonstrate that toxicity can be decoupled from antitumor activity in preclinical models by limiting IL2 signaling to CD8+ T cells, supporting the development of CD8+ T cell-selective IL2 for the treatment of cancer. See related article by Kaptein et al. p. 1226.
©2024 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
K.D. Moynihan reports other support from Asher Biotherapeutics during the conduct of the study; in addition, K.D. Moynihan has a patent for CD8 targeted IL2 pending. D.C. Pappas reports a patent for US20220162314A1 pending; and being a stock-holding employee of Asher Biotherapeutics. S. Chin reports a patent for US20220162314A1 pending to Asher Biotherapeutics Inc. R.Y. Lan reports other support from Asher Biotherapeutics during the conduct of the study; other support from Asher Biotherapeutics outside the submitted work. H.C. Nguyen reports a patent for US20220162314A1 pending. W. Chen reports a patent for US20220162314A1 pending. T.N. Schumacher reports personal fees from Asher Bio during the conduct of the study; personal fees from Neogene Therapeutics, Merus, Allogene Therapeutics, and Scenic Biotech outside the submitted work. R.D. Schreiber reports grants, personal fees, and other support from Asher Biotherapeutics during the conduct of the study; personal fees and other support from A2 Biotherapeutics, grants, personal fees, and other support from NGM Biotherapeutics, Sensei Biotherapeutics, and personal fees from BlueSphere Bio outside the submitted work. Y.A. Yeung reports a patent for US20220162314A1 pending and a patent for US20220251202A1 pending. I.M. Djuretic reports other support from Asher Biotherapeutics during the conduct of the study; other support from Asher Biotherapeutics outside the submitted work; in addition, I.M. Djuretic has a patent for CD8-targeted IL2 molecules pending to Asher Biotherapeutics. No disclosures were reported by the other authors.
Figures

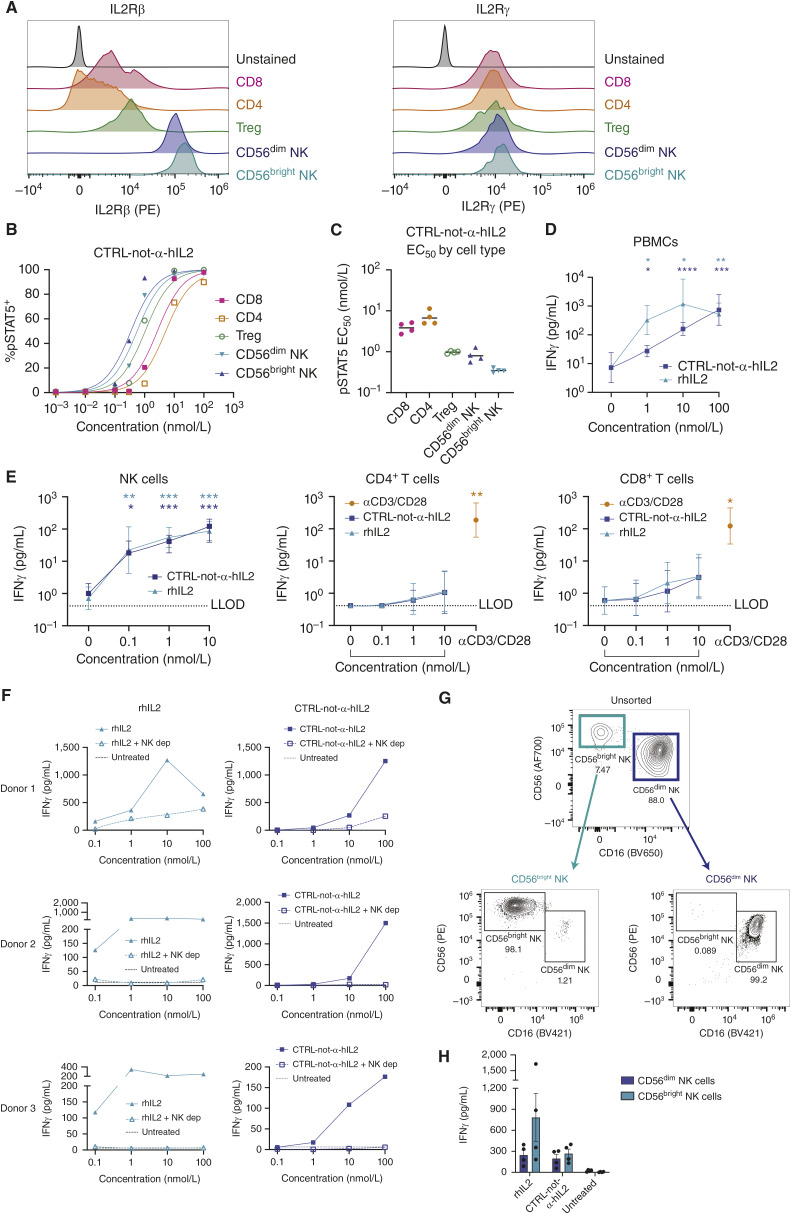





References
-
- Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, et al. . Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA 1994;271:907. - PubMed
-
- Chatzkel J, Schell MJ, Chahoud J, Zhang J, Jain R, Swank J, et al. . Coordinated pembrolizumab and high dose IL-2 (5-in-a-row schedule) for therapy of metastatic clear cell renal cancer. Clin Genitourin Cancer 2022;20:252–9. - PubMed
-
- Abbas AK, Trotta E, Simeonov DR, Marson A, Bluestone JA. Revisiting IL-2: biology and therapeutic prospects. Sci Immunol 2018;3:eaat1482. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
