

Characterization of the transcriptionally active form of dephosphorylated DctD complexed with dephospho-IIAGlc
- PMID: 38564689
- PMCID: PMC11077940
- DOI: 10.1128/mbio.00330-24
Characterization of the transcriptionally active form of dephosphorylated DctD complexed with dephospho-IIAGlc
Abstract
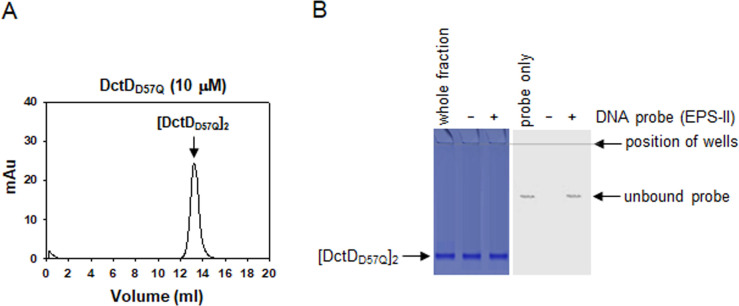
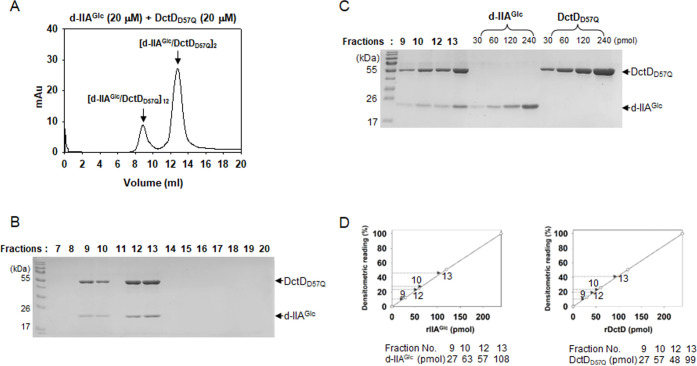
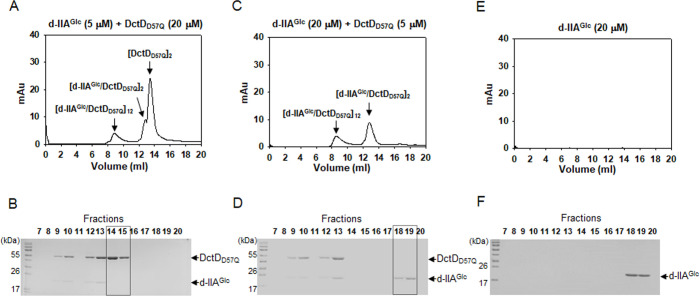
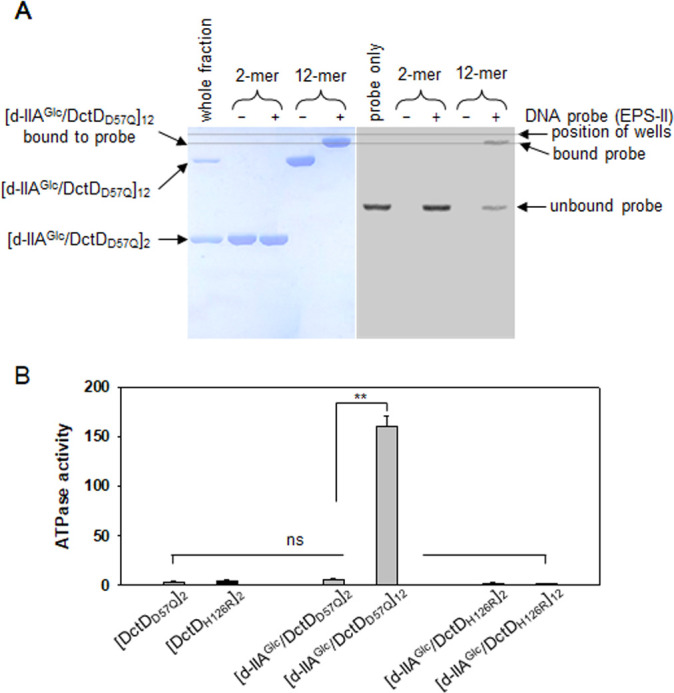
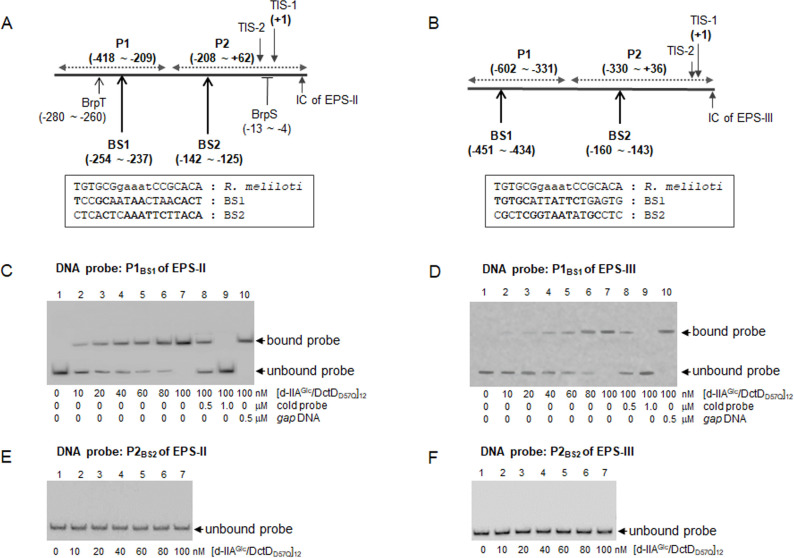
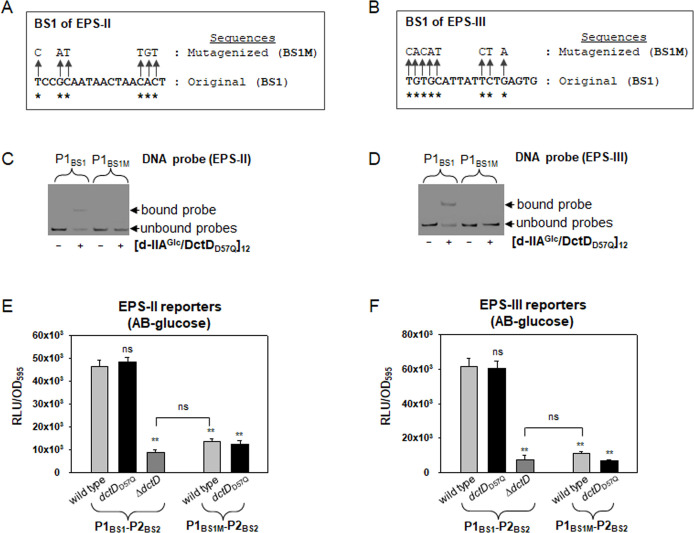
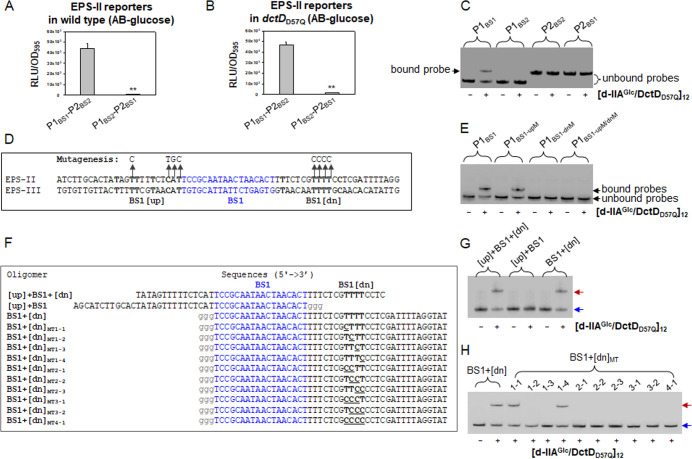
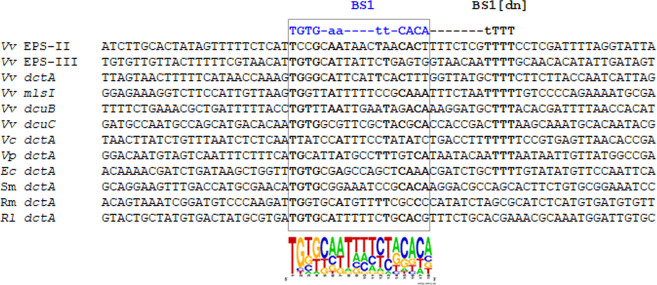
Bacterial enhancer-binding proteins (bEBPs) acquire a transcriptionally active state via phosphorylation. However, transcriptional activation by the dephosphorylated form of bEBP has been observed in DctD, which belongs to Group I bEBP. The formation of a complex between dephosphorylated DctD (d-DctD) and dephosphorylated IIAGlc (d-IIAGlc) is a prerequisite for the transcriptional activity of d-DctD. In the present study, characteristics of the transcriptionally active complex composed of d-IIAGlc and phosphorylation-deficient DctD (DctDD57Q) of Vibrio vulnificus were investigated in its multimeric conformation and DNA-binding ability. DctDD57Q formed a homodimer that could not bind to the DNA. In contrast, when DctDD57Q formed a complex with d-IIAGlc in a 1:1 molar ratio, it produced two conformations: dimer and dodecamer of the complex. Only the dodecameric complex exhibited ATP-hydrolyzing activity and DNA-binding affinity. For successful DNA-binding and transcriptional activation by the dodecameric d-IIAGlc/DctDD57Q complex, extended upstream activator sequences were required, which encompass the nucleotide sequences homologous to the known DctD-binding site and additional nucleotides downstream. This is the first report to demonstrate the molecular characteristics of a dephosphorylated bEBP complexed with another protein to form a transcriptionally active dodecameric complex, which has an affinity for a specific DNA-binding sequence.IMPORTANCEResponse regulators belonging to the bacterial two-component regulatory system activate the transcription initiation of their regulons when they are phosphorylated by cognate sensor kinases and oligomerized to the appropriate multimeric states. Recently, it has been shown that a dephosphorylated response regulator, DctD, could activate transcription in a phosphorylation-independent manner in Vibrio vulnificus. The dephosphorylated DctD activated transcription as efficiently as phosphorylated DctD when it formed a complex with dephosphorylated form of IIAGlc, a component of the glucose-phosphotransferase system. Functional mimicry of this complex with the typical form of transcriptionally active phosphorylated DctD led us to study the molecular characteristics of this heterodimeric complex. Through systematic analyses, it was surprisingly determined that a multimer constituted with 12 complexes gained the ability to hydrolyze ATP and recognize specific upstream activator sequences containing a typical inverted-repeat sequence flanked by distinct nucleotides.
Keywords: bacterial enhancer-binding protein; dephospho-DctD/dephospho-IIAGlccomplex; dodecameric conformation; upstream activator sequences.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Golby P, Davies S, Kelly DJ, Guest JR, Andrews SC. 1999. Identification and characterization of a two-component sensor-kinase and response-regulator system (DcuS-DcuR) controlling gene expression in response to C4-dicarboxylates in Escherichia coli. J Bacteriol 181:1238–1248. doi:10.1128/JB.181.4.1238-1248.1999 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
