

Early detection of emerging viral variants through analysis of community structure of coordinated substitution networks
- PMID: 38565543
- PMCID: PMC10987511
- DOI: 10.1038/s41467-024-47304-6
Early detection of emerging viral variants through analysis of community structure of coordinated substitution networks
Abstract
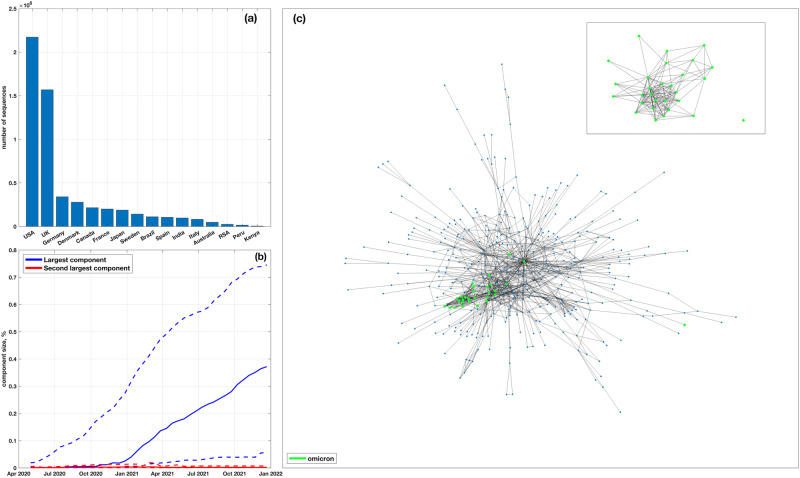
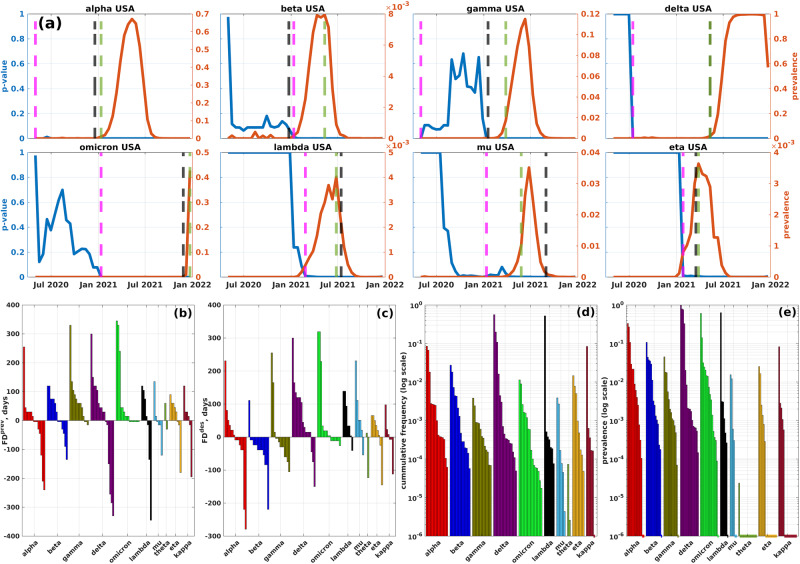
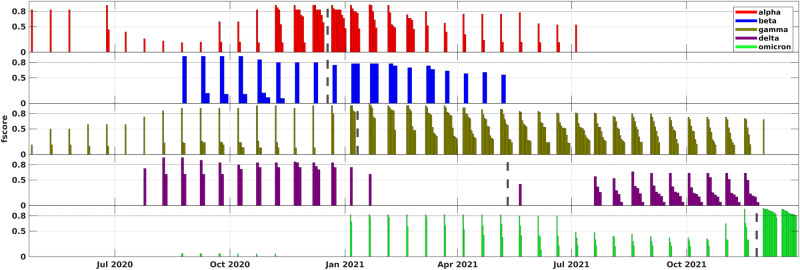
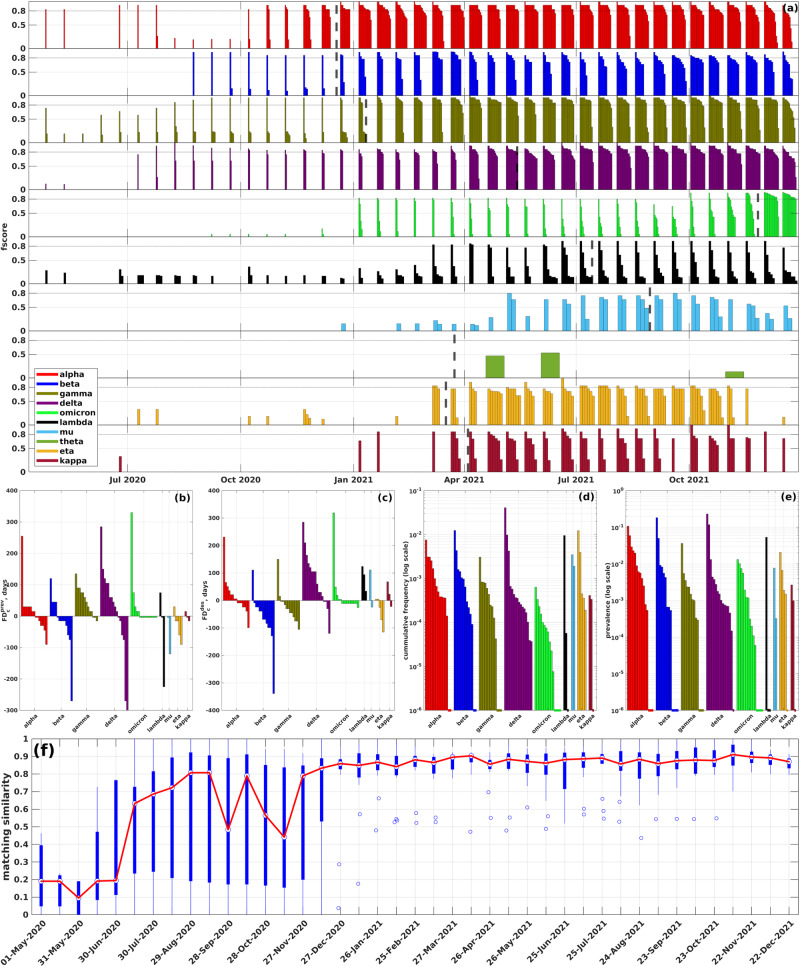
The emergence of viral variants with altered phenotypes is a public health challenge underscoring the need for advanced evolutionary forecasting methods. Given extensive epistatic interactions within viral genomes and known viral evolutionary history, efficient genomic surveillance necessitates early detection of emerging viral haplotypes rather than commonly targeted single mutations. Haplotype inference, however, is a significantly more challenging problem precluding the use of traditional approaches. Here, using SARS-CoV-2 evolutionary dynamics as a case study, we show that emerging haplotypes with altered transmissibility can be linked to dense communities in coordinated substitution networks, which become discernible significantly earlier than the haplotypes become prevalent. From these insights, we develop a computational framework for inference of viral variants and validate it by successful early detection of known SARS-CoV-2 strains. Our methodology offers greater scalability than phylogenetic lineage tracing and can be applied to any rapidly evolving pathogen with adequate genomic surveillance data.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
TopHap: rapid inference of key phylogenetic structures from common haplotypes in large genome collections with limited diversity.Bioinformatics. 2022 May 13;38(10):2719-2726. doi: 10.1093/bioinformatics/btac186. Bioinformatics. 2022. PMID: 35561179 Free PMC article.
-
Comparative Genomics Reveals Early Emergence and Biased Spatiotemporal Distribution of SARS-CoV-2.Mol Biol Evol. 2021 May 19;38(6):2547-2565. doi: 10.1093/molbev/msab049. Mol Biol Evol. 2021. PMID: 33605421 Free PMC article.
-
Haplotype distribution of SARS-CoV-2 variants in low and high vaccination rate countries during ongoing global COVID-19 pandemic in early 2021.Infect Genet Evol. 2022 Jan;97:105164. doi: 10.1016/j.meegid.2021.105164. Epub 2021 Nov 27. Infect Genet Evol. 2022. PMID: 34848355 Free PMC article.
-
Global SARS-CoV-2 genomic surveillance: What we have learned (so far).Infect Genet Evol. 2023 Mar;108:105405. doi: 10.1016/j.meegid.2023.105405. Epub 2023 Jan 18. Infect Genet Evol. 2023. PMID: 36681102 Free PMC article. Review.
-
Evolutionary study of COVID-19, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) as an emerging coronavirus: Phylogenetic analysis and literature review.Vet Med Sci. 2021 Mar;7(2):559-571. doi: 10.1002/vms3.394. Epub 2020 Nov 18. Vet Med Sci. 2021. PMID: 33210477 Free PMC article. Review.
Cited by
-
Epidemiological inference at the threshold of data availability: an influenza A(H1N2)v spillover event in the United Kingdom.J R Soc Interface. 2024 Aug;21(217):20240168. doi: 10.1098/rsif.2024.0168. Epub 2024 Aug 7. J R Soc Interface. 2024. PMID: 39109454 Free PMC article.
-
Identification of dynamic networks community by fusing deep learning and evolutionary clustering.Sci Rep. 2024 Oct 10;14(1):23741. doi: 10.1038/s41598-024-74361-0. Sci Rep. 2024. PMID: 39390015 Free PMC article.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
