

A concerted increase in readthrough and intron retention drives transposon expression during aging and senescence
- PMID: 38567944
- PMCID: PMC10990488
- DOI: 10.7554/eLife.87811
A concerted increase in readthrough and intron retention drives transposon expression during aging and senescence
Abstract
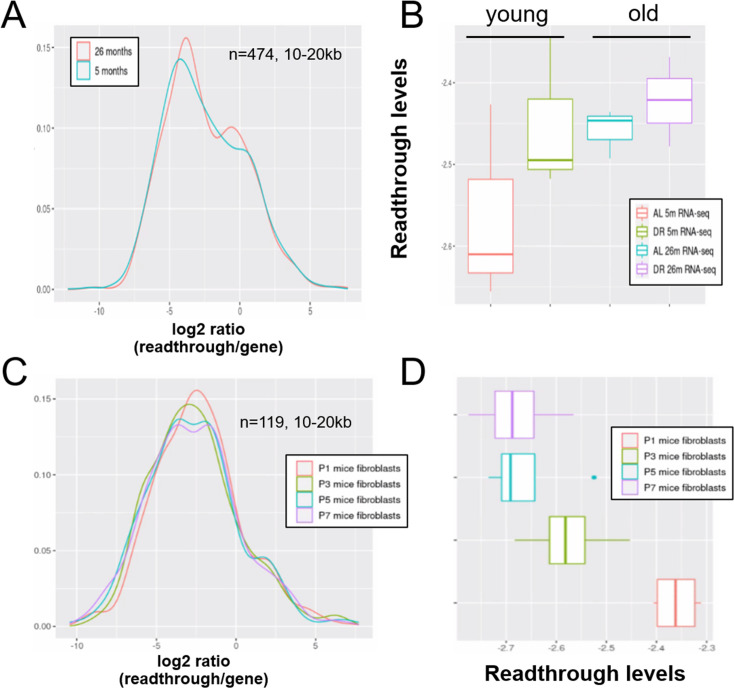
Aging and senescence are characterized by pervasive transcriptional dysfunction, including increased expression of transposons and introns. Our aim was to elucidate mechanisms behind this increased expression. Most transposons are found within genes and introns, with a large minority being close to genes. This raises the possibility that transcriptional readthrough and intron retention are responsible for age-related changes in transposon expression rather than expression of autonomous transposons. To test this, we compiled public RNA-seq datasets from aged human fibroblasts, replicative and drug-induced senescence in human cells, and RNA-seq from aging mice and senescent mouse cells. Indeed, our reanalysis revealed a correlation between transposons expression, intron retention, and transcriptional readthrough across samples and within samples. Both intron retention and readthrough increased with aging or cellular senescence and these transcriptional defects were more pronounced in human samples as compared to those of mice. In support of a causal connection between readthrough and transposon expression, analysis of models showing induced transcriptional readthrough confirmed that they also show elevated transposon expression. Taken together, our data suggest that elevated transposon reads during aging seen in various RNA-seq dataset are concomitant with multiple transcriptional defects. Intron retention and transcriptional readthrough are the most likely explanation for the expression of transposable elements that lack a functional promoter.
Keywords: aging; chromosomes; gene expression; human; intron retention; mouse; senescense; transcriptional readthrough; transposon expression.
© 2023, Pabis et al.
Conflict of interest statement
KP, DB, OS, KS, JG, BK No competing interests declared
Figures





































Update of
- doi: 10.1101/2022.12.29.522277
- doi: 10.7554/eLife.87811.1
- doi: 10.7554/eLife.87811.2
References
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
