

Arsenic trioxide impacts hepatitis B virus core nuclear localization and efficiently interferes with HBV infection
- PMID: 38567974
- PMCID: PMC11064512
- DOI: 10.1128/spectrum.03788-23
Arsenic trioxide impacts hepatitis B virus core nuclear localization and efficiently interferes with HBV infection
Abstract
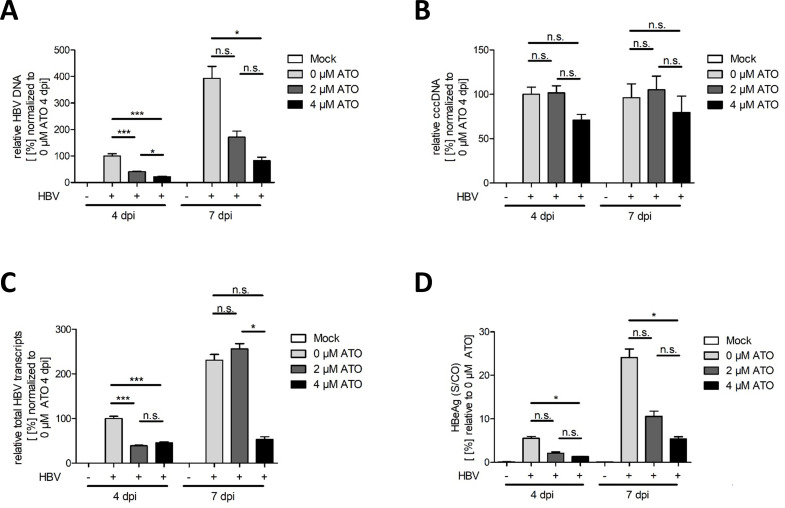
The key to a curative treatment of hepatitis B virus (HBV) infection is the eradication of the intranuclear episomal covalently closed circular DNA (cccDNA), the stable persistence reservoir of HBV. Currently, established therapies can only limit HBV replication but fail to tackle the cccDNA. Thus, novel therapeutic approaches toward curative treatment are urgently needed. Recent publications indicated a strong association between the HBV core protein SUMOylation and the association with promyelocytic leukemia nuclear bodies (PML-NBs) on relaxed circular DNA to cccDNA conversion. We propose that interference with the cellular SUMOylation system and PML-NB integrity using arsenic trioxide provides a useful tool in the treatment of HBV infection. Our study showed a significant reduction in HBV-infected cells, core protein levels, HBV mRNA, and total DNA. Additionally, a reduction, albeit to a limited extent, of HBV cccDNA could be observed. Furthermore, this interference was also applied for the treatment of an established HBV infection, characterized by a stably present nuclear pool of cccDNA. Arsenic trioxide (ATO) treatment not only changed the amount of expressed HBV core protein but also induced a distinct relocalization to an extranuclear phenotype during infection. Moreover, ATO treatment resulted in the redistribution of transfected HBV core protein away from PML-NBs, a phenotype similar to that previously observed with SUMOylation-deficient HBV core. Taken together, these findings revealed the inhibition of HBV replication by ATO treatment during several steps of the viral replication cycle, including viral entry into the nucleus as well as cccDNA formation and maintenance. We propose ATO as a novel prospective treatment option for further pre-clinical and clinical studies against HBV infection.
Importance: The main challenge for the achievement of a functional cure for hepatitis B virus (HBV) is the covalently closed circular DNA (cccDNA), the highly stable persistence reservoir of HBV, which is maintained by further rounds of infection with newly generated progeny viruses or by intracellular recycling of mature nucleocapsids. Eradication of the cccDNA is considered to be the holy grail for HBV curative treatment; however, current therapeutic approaches fail to directly tackle this HBV persistence reservoir. The molecular effect of arsenic trioxide (ATO) on HBV infection, protein expression, and cccDNA formation and maintenance, however, has not been characterized and understood until now. In this study, we reveal ATO treatment as a novel and innovative therapeutic approach against HBV infections, repressing viral gene expression and replication as well as the stable cccDNA pool at low micromolar concentrations by affecting the cellular function of promyelocytic leukemia nuclear bodies.
Keywords: HBV; PML-NB; SUMO; antivirals; arsenic; cccDNA; dsDNA viruses; hepatitis B virus.
Conflict of interest statement
U.P. is a co-founder and shareholder of SCG Cell Therapy and received personal fees as a speaker or an
Figures







References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
