

Homocysteine-induced sustained GluN2A NMDA receptor stimulation leads to mitochondrial ROS generation and neurotoxicity
- PMID: 38569938
- PMCID: PMC11081806
- DOI: 10.1016/j.jbc.2024.107253
Homocysteine-induced sustained GluN2A NMDA receptor stimulation leads to mitochondrial ROS generation and neurotoxicity
Abstract
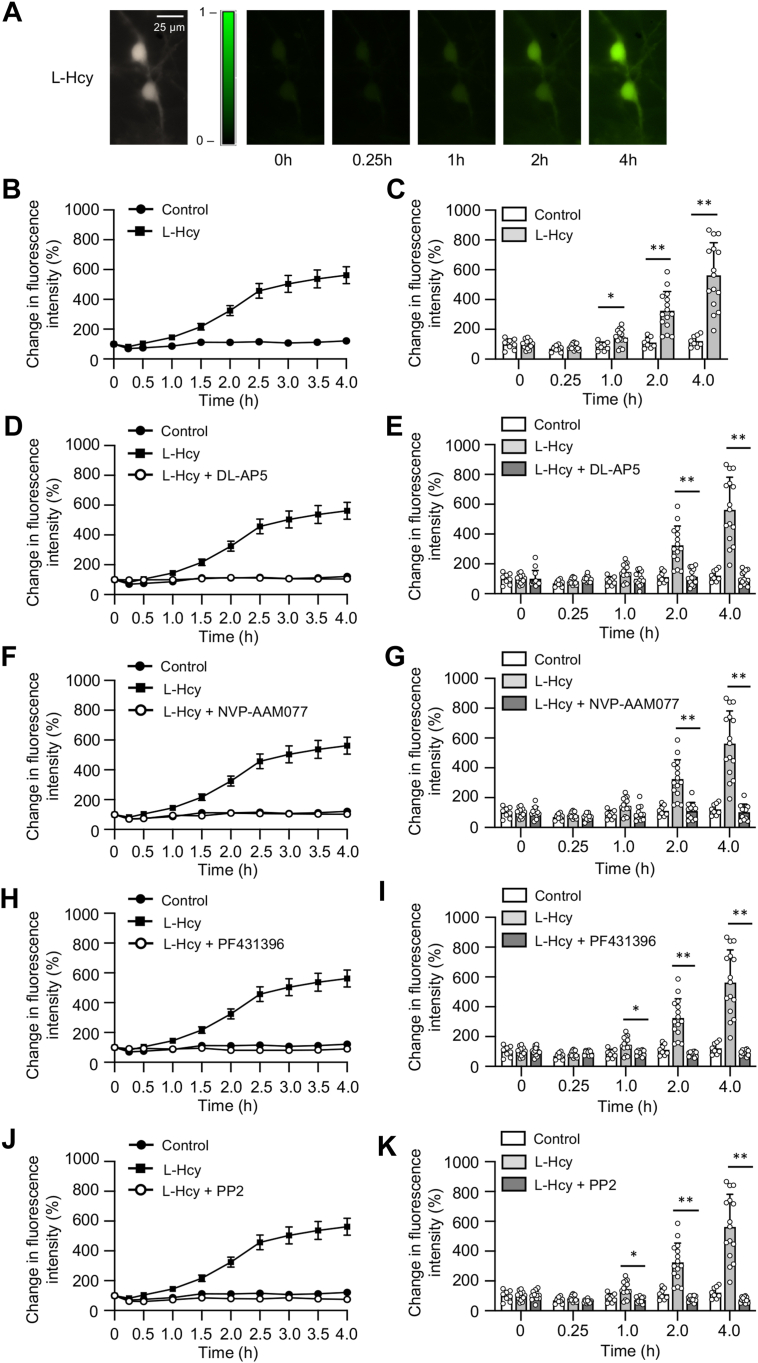
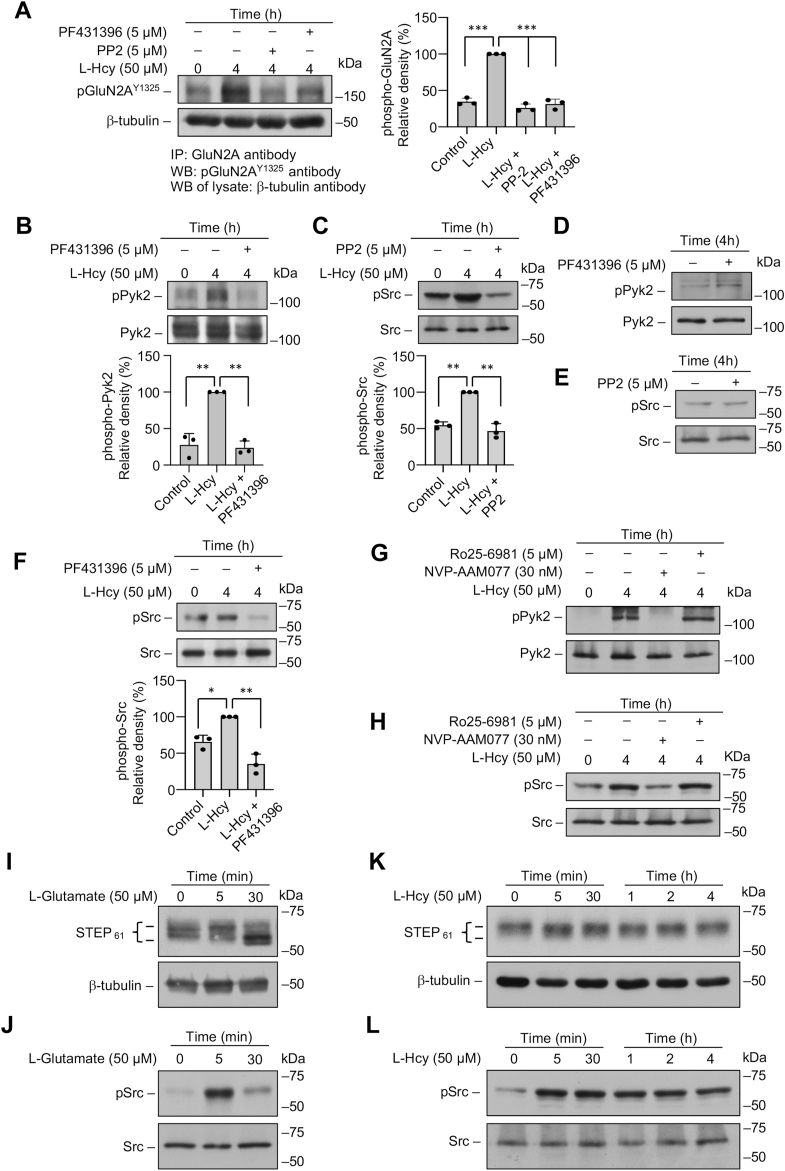
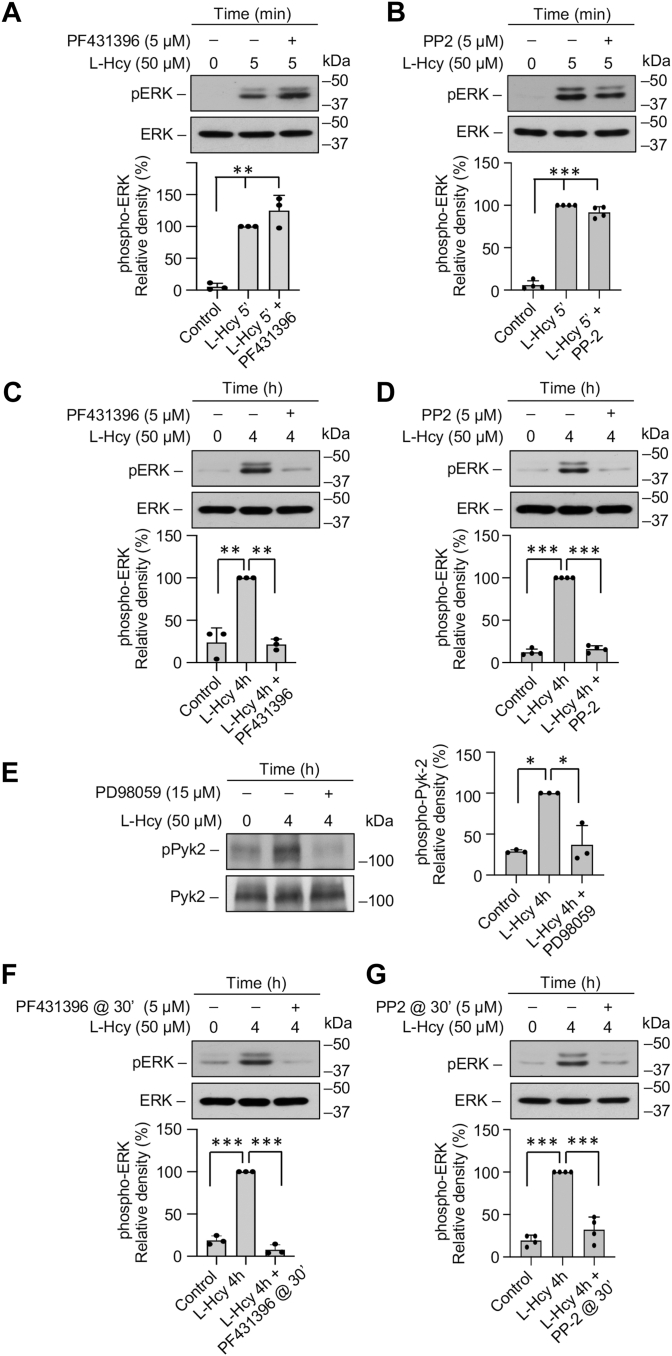
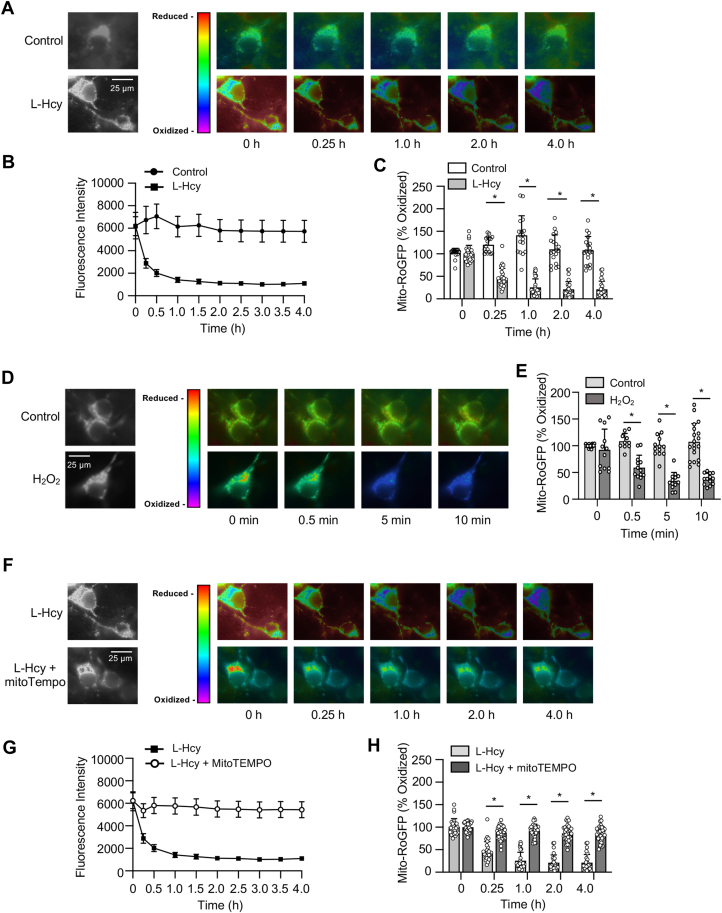
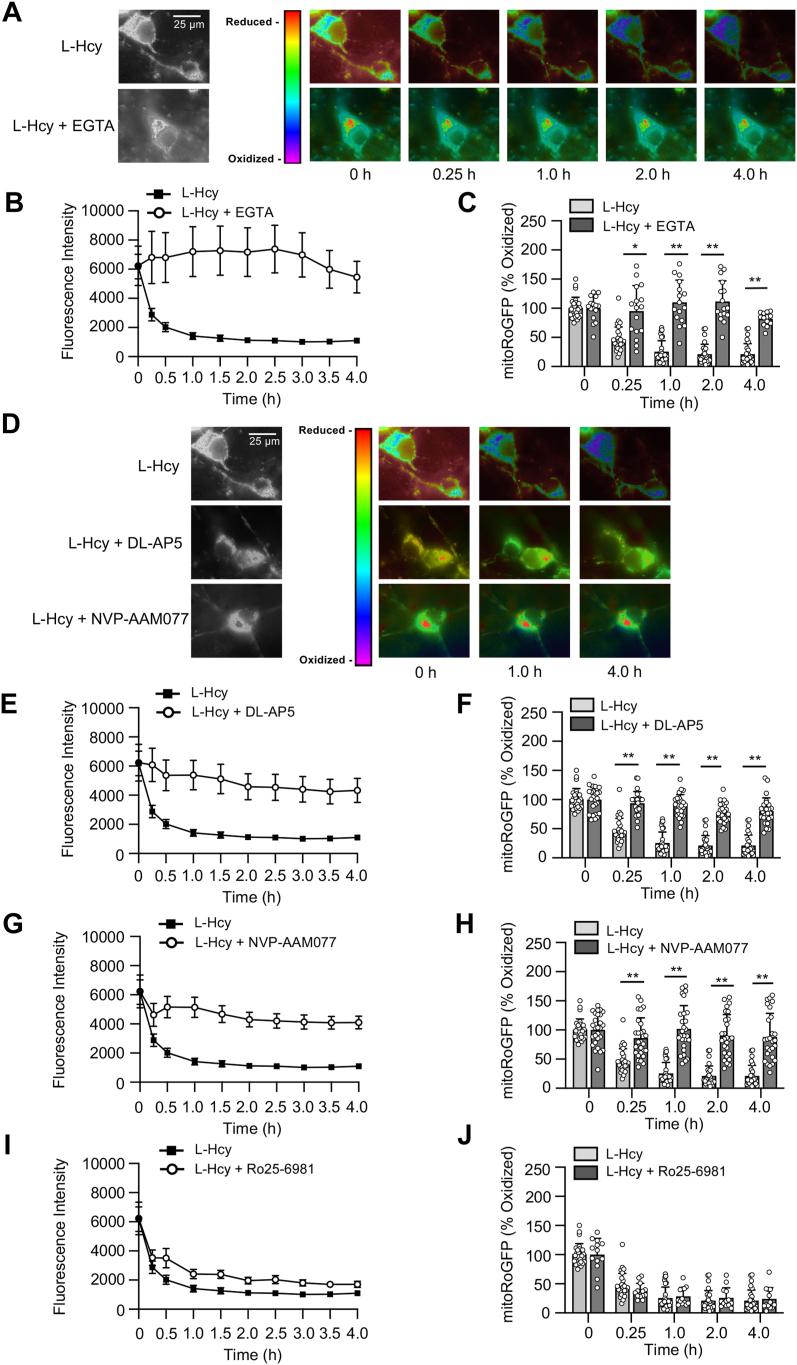
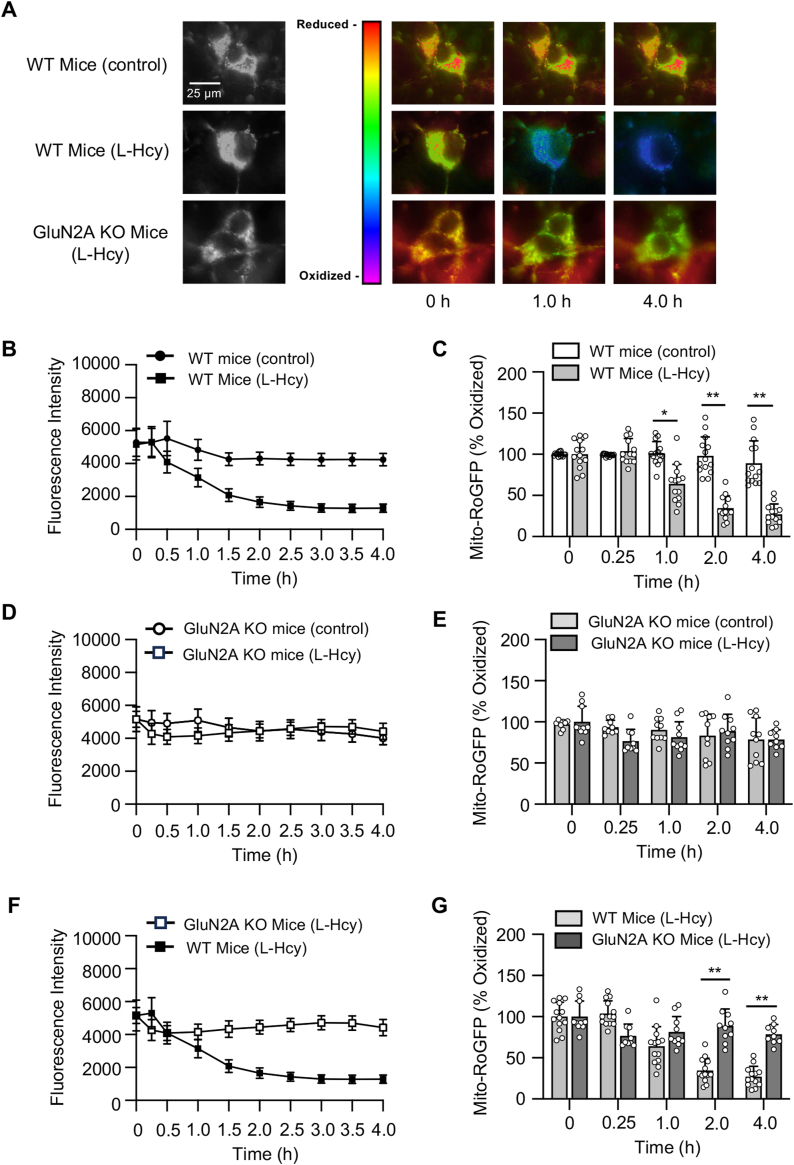
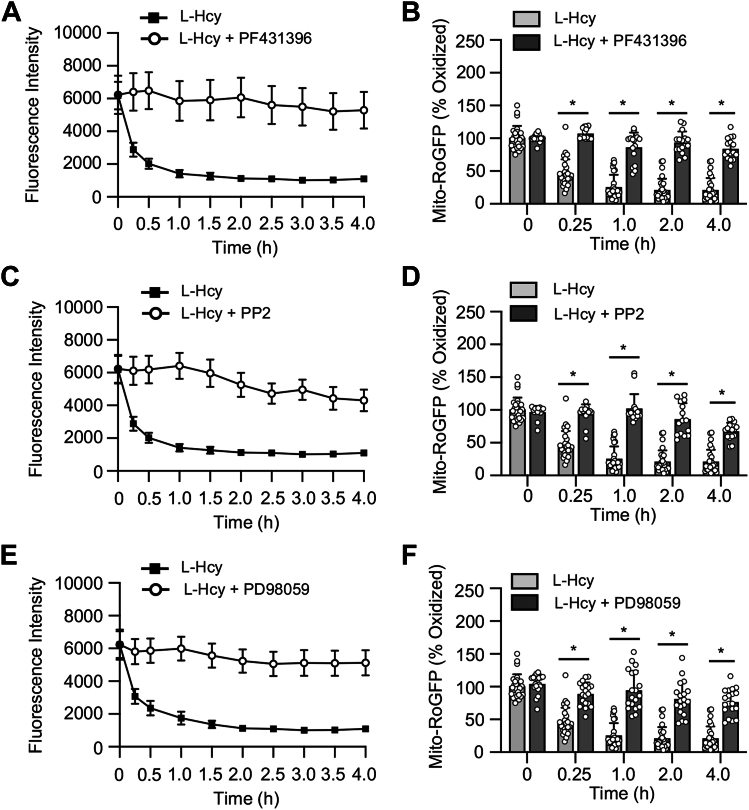
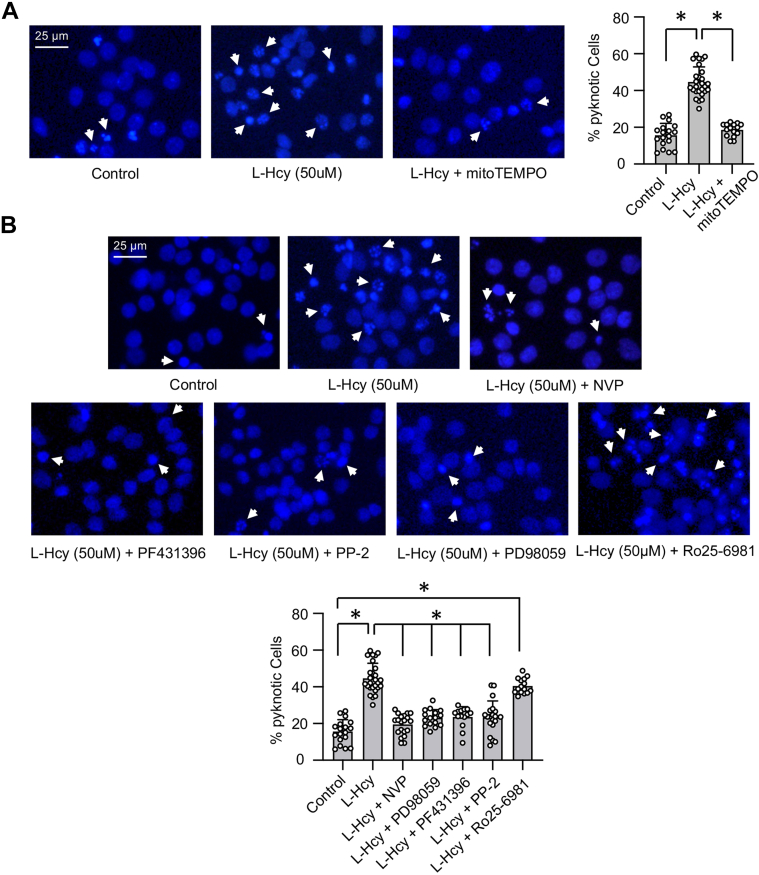
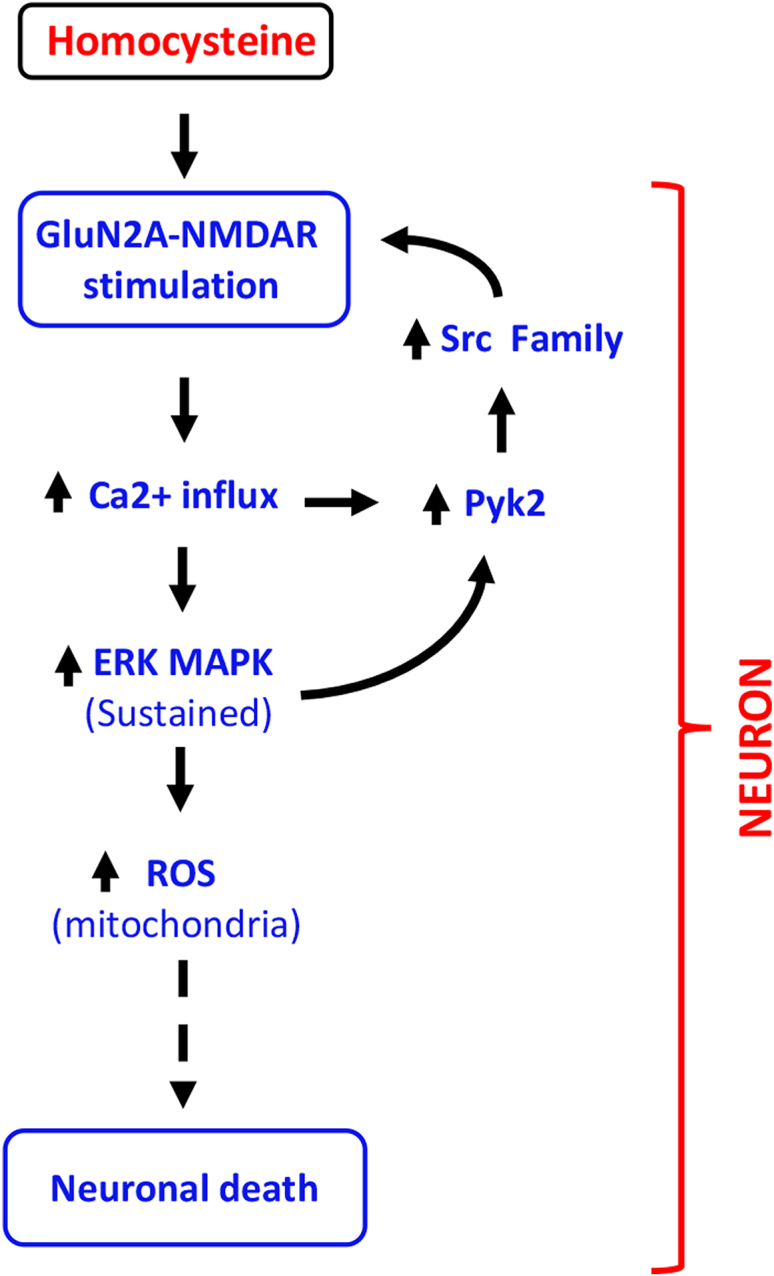
Homocysteine, a sulfur-containing amino acid derived from methionine metabolism, is a known agonist of N-methyl-D-aspartate receptor (NMDAR) and is involved in neurotoxicity. Our previous findings showed that neuronal exposure to elevated homocysteine levels leads to sustained low-level increase in intracellular Ca2+, which is dependent on GluN2A subunit-containing NMDAR (GluN2A-NMDAR) stimulation. These studies further showed a role of ERK MAPK in homocysteine-GluN2A-NMDAR-mediated neuronal death. However, the intracellular mechanisms associated with such sustained GluN2A-NMDAR stimulation and subsequent Ca2+ influx have remained unexplored. Using live-cell imaging with Fluo3-AM and biochemical approaches, we show that homocysteine-GluN2A NMDAR-induced initial Ca2+ influx triggers sequential phosphorylation and subsequent activation of the proline rich tyrosine kinase 2 (Pyk2) and Src family kinases, which in turn phosphorylates GluN2A-Tyr1325 residue of GluN2A-NMDARs to maintain channel activity. The continuity of this cycle of events leads to sustained Ca2+ influx through GluN2A-NMDAR. Our findings also show that lack of activation of the regulatory tyrosine phosphatase STEP, which can limit Pyk2 and Src family kinase activity further contributes to the maintenance of this cycle. Additional studies using live-cell imaging of neurons expressing a redox-sensitive GFP targeted to the mitochondrial matrix show that treatment with homocysteine leads to a progressive increase in mitochondrial reactive oxygen species generation, which is dependent on GluN2A-NMDAR-mediated sustained ERK MAPK activation. This later finding demonstrates a novel role of GluN2A-NMDAR in homocysteine-induced mitochondrial ROS generation and highlights the role of ERK MAPK as the intermediary signaling pathway between GluN2A-NMDAR stimulation and mitochondrial reactive oxygen species generation.
Keywords: Ca(2+) influx; ERK MAPK; GluN2A-NMDA receptor; Pyk2; Src family kinase; homocysteine; mitochondrial reactive oxygen species; neurotoxicity.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures
References
-
- Hardingham G.E., Fukunaga Y., Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002;5:405–414. - PubMed
-
- Kim M.J., Dunah A.W., Wang Y.T., Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. - PubMed
-
- Li B., Chen N., Luo T., Otsu Y., Murphy T.H., Raymond L.A. Differential regulation of synaptic and extra-synaptic NMDA receptors. Nat. Neurosci. 2002;5:833–834. - PubMed
-
- Zhang S.J., Steijaert M.N., Lau D., Schutz G., Delucinge-Vivier C., Descombes P., et al. Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron. 2007;53:549–562. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
