

Genomic language model predicts protein co-regulation and function
- PMID: 38570504
- PMCID: PMC10991518
- DOI: 10.1038/s41467-024-46947-9
Genomic language model predicts protein co-regulation and function
Abstract
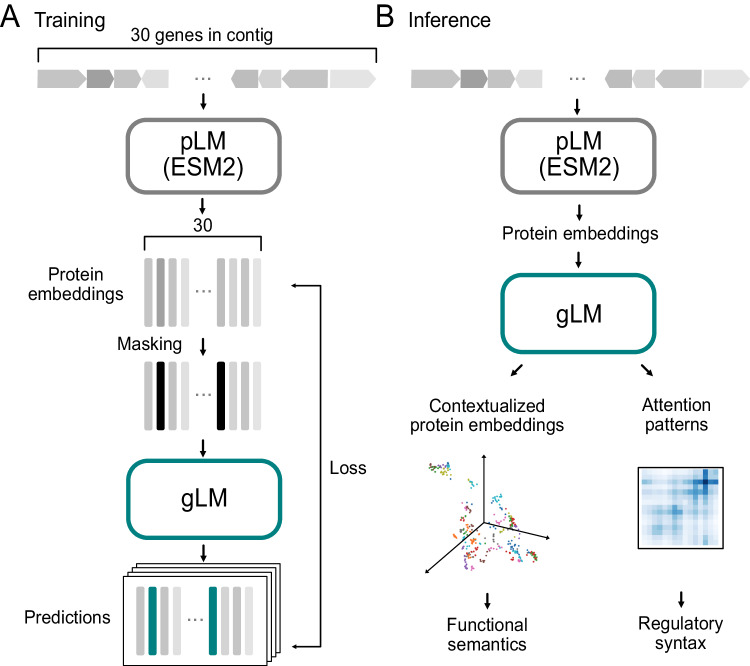
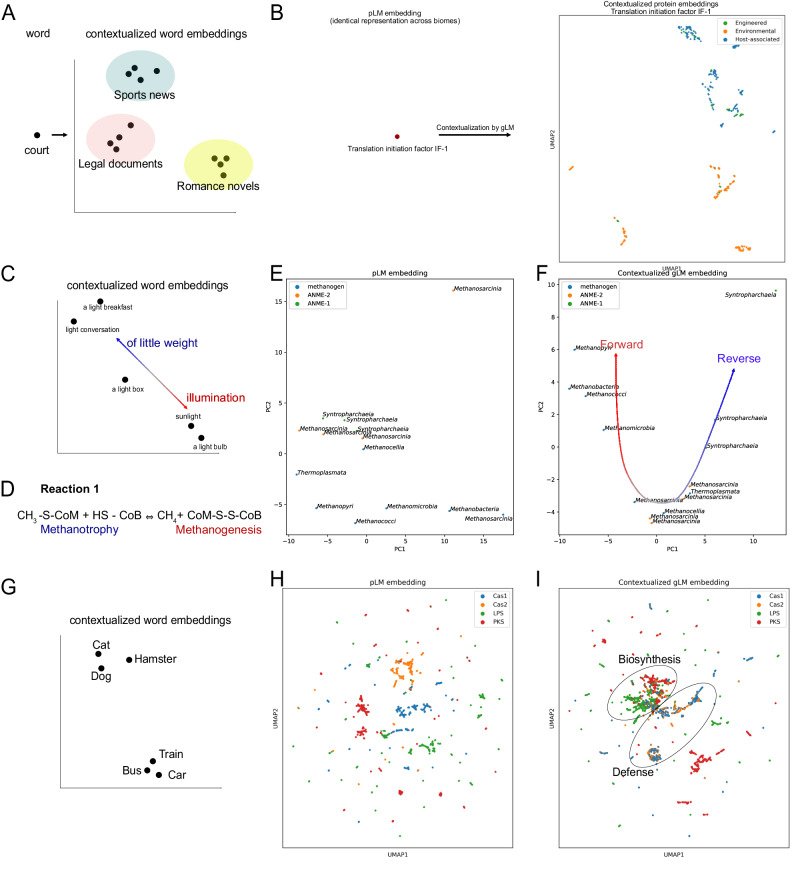
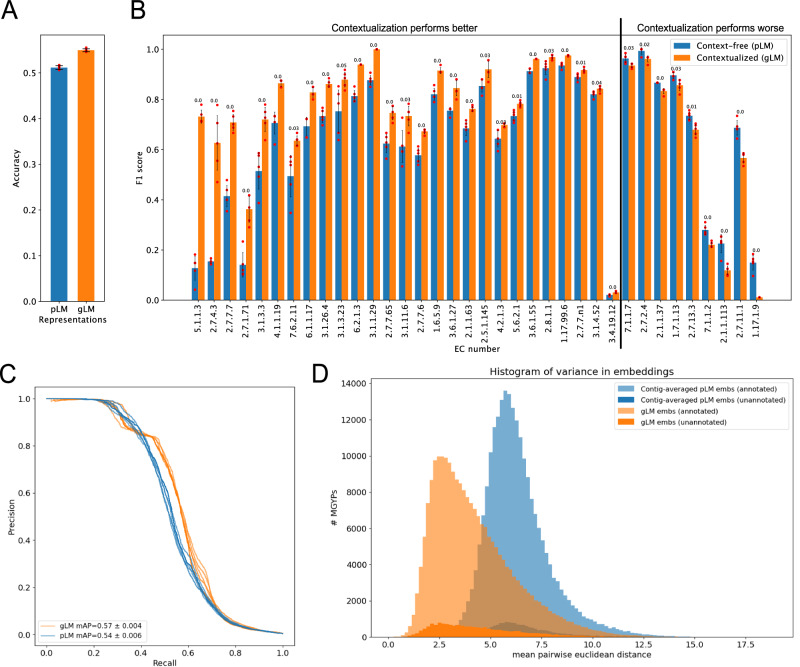
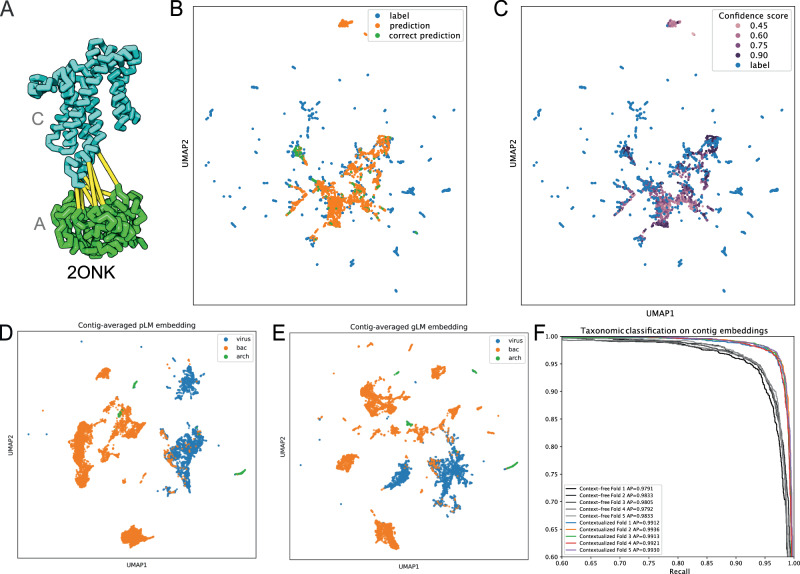
Deciphering the relationship between a gene and its genomic context is fundamental to understanding and engineering biological systems. Machine learning has shown promise in learning latent relationships underlying the sequence-structure-function paradigm from massive protein sequence datasets. However, to date, limited attempts have been made in extending this continuum to include higher order genomic context information. Evolutionary processes dictate the specificity of genomic contexts in which a gene is found across phylogenetic distances, and these emergent genomic patterns can be leveraged to uncover functional relationships between gene products. Here, we train a genomic language model (gLM) on millions of metagenomic scaffolds to learn the latent functional and regulatory relationships between genes. gLM learns contextualized protein embeddings that capture the genomic context as well as the protein sequence itself, and encode biologically meaningful and functionally relevant information (e.g. enzymatic function, taxonomy). Our analysis of the attention patterns demonstrates that gLM is learning co-regulated functional modules (i.e. operons). Our findings illustrate that gLM's unsupervised deep learning of the metagenomic corpus is an effective and promising approach to encode functional semantics and regulatory syntax of genes in their genomic contexts and uncover complex relationships between genes in a genomic region.
© 2024. The Author(s).
Conflict of interest statement
A provisional patent (App. Serial No.: 63/491,019) on this work was filed by Harvard University with YH and SO as inventors. The remaining authors declare no competing interests.
Figures

Similar articles
-
FGeneBERT: function-driven pre-trained gene language model for metagenomics.Brief Bioinform. 2025 Mar 4;26(2):bbaf149. doi: 10.1093/bib/bbaf149. Brief Bioinform. 2025. PMID: 40211978 Free PMC article.
-
A comparison of word embeddings for the biomedical natural language processing.J Biomed Inform. 2018 Nov;87:12-20. doi: 10.1016/j.jbi.2018.09.008. Epub 2018 Sep 12. J Biomed Inform. 2018. PMID: 30217670 Free PMC article.
-
Operons and the effect of genome redundancy in deciphering functional relationships using phylogenetic profiles.Proteins. 2008 Feb 1;70(2):344-52. doi: 10.1002/prot.21564. Proteins. 2008. PMID: 17671982
-
Gene context conservation of a higher order than operons.Trends Biochem Sci. 2000 Oct;25(10):474-9. doi: 10.1016/s0968-0004(00)01663-7. Trends Biochem Sci. 2000. PMID: 11050428 Review.
-
Predicting Functional Interactions Among Genes in Prokaryotes by Genomic Context.Adv Exp Med Biol. 2015;883:97-106. doi: 10.1007/978-3-319-23603-2_5. Adv Exp Med Biol. 2015. PMID: 26621463 Review.
Cited by
-
Protein Set Transformer: A protein-based genome language model to power high diversity viromics.Res Sq [Preprint]. 2024 Sep 23:rs.3.rs-4844047. doi: 10.21203/rs.3.rs-4844047/v1. Res Sq. 2024. PMID: 39399683 Free PMC article. Preprint.
-
PlasGO: enhancing GO-based function prediction for plasmid-encoded proteins based on genetic structure.Gigascience. 2024 Jan 2;13:giae104. doi: 10.1093/gigascience/giae104. Gigascience. 2024. PMID: 39704702 Free PMC article.
-
Protein Sequence Analysis landscape: A Systematic Review of Task Types, Databases, Datasets, Word Embeddings Methods, and Language Models.Database (Oxford). 2025 May 30;2025:baaf027. doi: 10.1093/database/baaf027. Database (Oxford). 2025. PMID: 40448683 Free PMC article.
-
AI-Driven Antimicrobial Peptide Discovery: Mining and Generation.Acc Chem Res. 2025 Jun 17;58(12):1831-1846. doi: 10.1021/acs.accounts.0c00594. Epub 2025 Jun 3. Acc Chem Res. 2025. PMID: 40459283 Free PMC article. Review.
-
Large-Scale Multi-omic Biosequence Transformers for Modeling Protein-Nucleic Acid Interactions.ArXiv [Preprint]. 2025 Jun 18:arXiv:2408.16245v5. ArXiv. 2025. PMID: 40236839 Free PMC article. Preprint.
References
-
- Elnaggar, A. et al. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112–7127 (2022). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
