

Mesenchymal glioma stem cells trigger vasectasia-distinct neovascularization process stimulated by extracellular vesicles carrying EGFR
- PMID: 38570528
- PMCID: PMC10991552
- DOI: 10.1038/s41467-024-46597-x
Mesenchymal glioma stem cells trigger vasectasia-distinct neovascularization process stimulated by extracellular vesicles carrying EGFR
Abstract
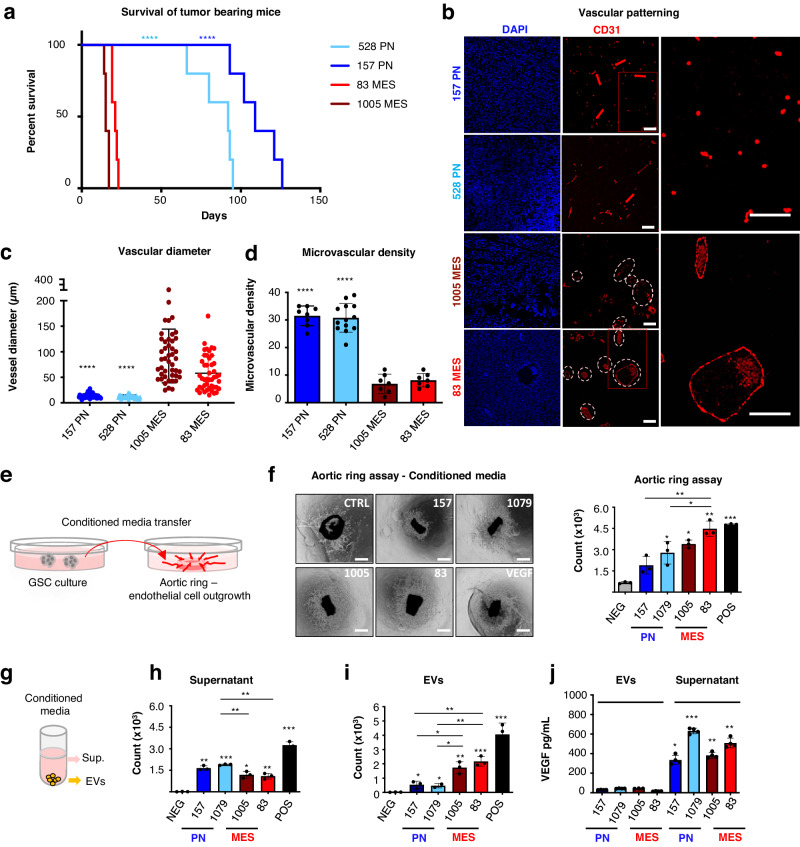
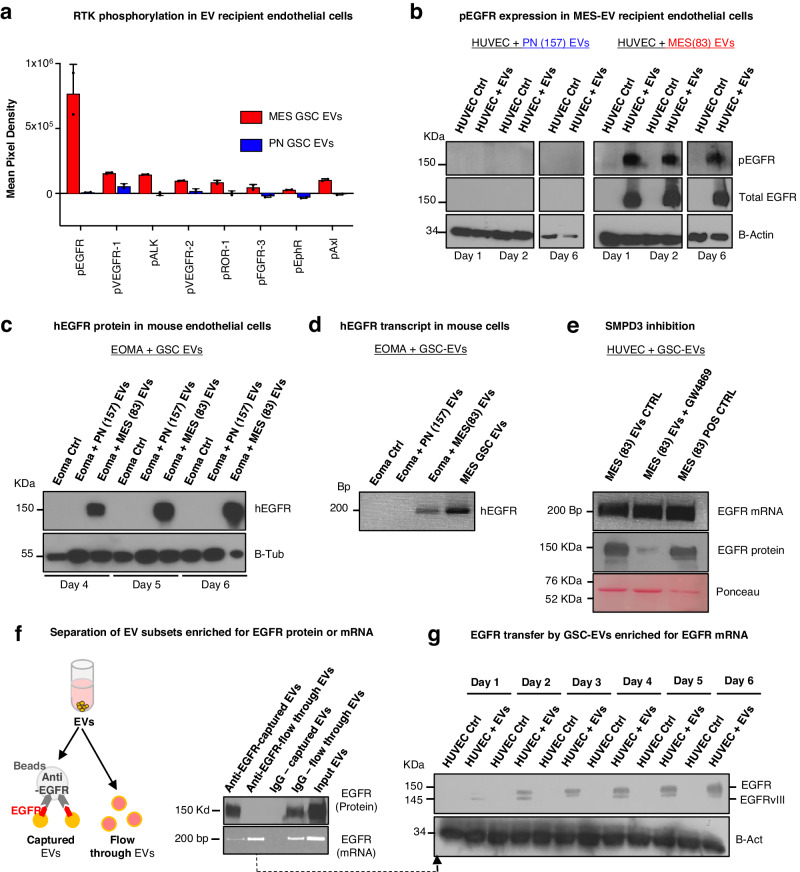
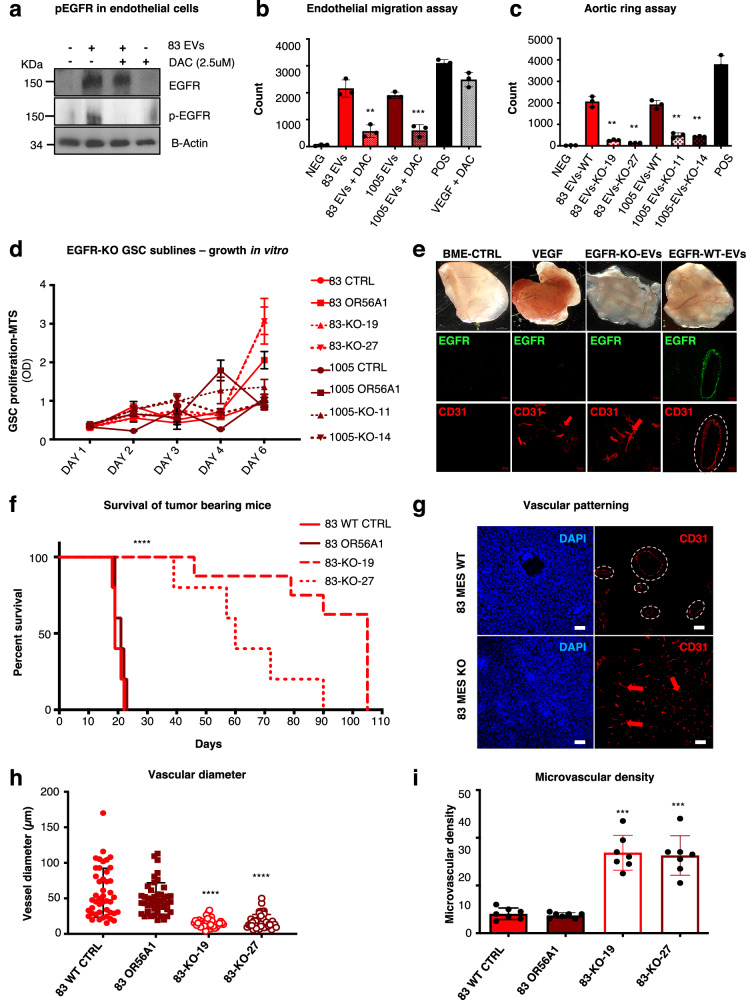
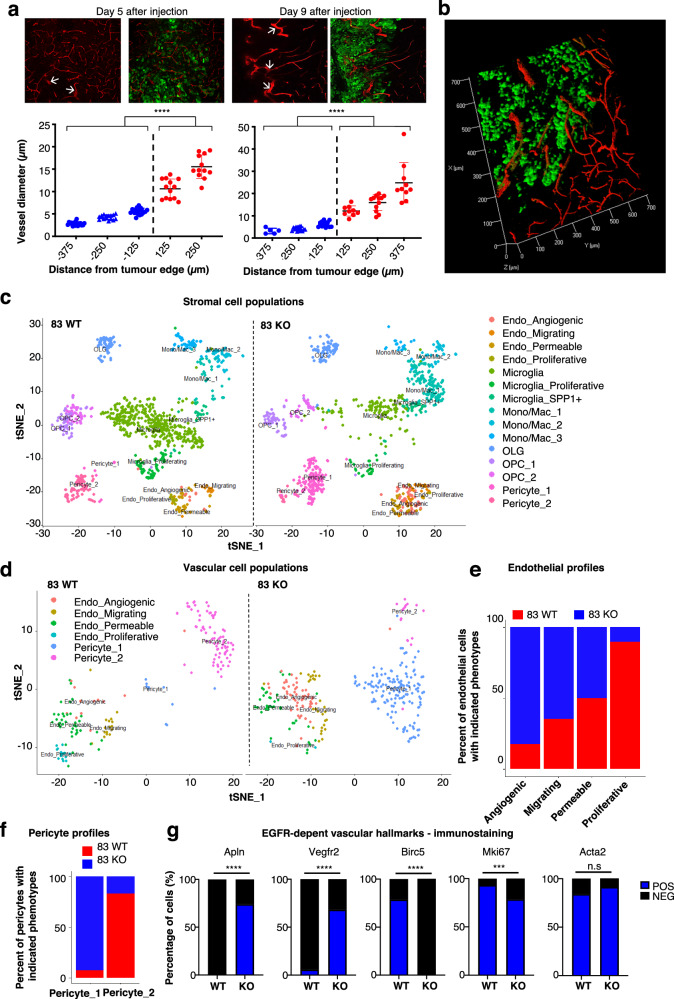
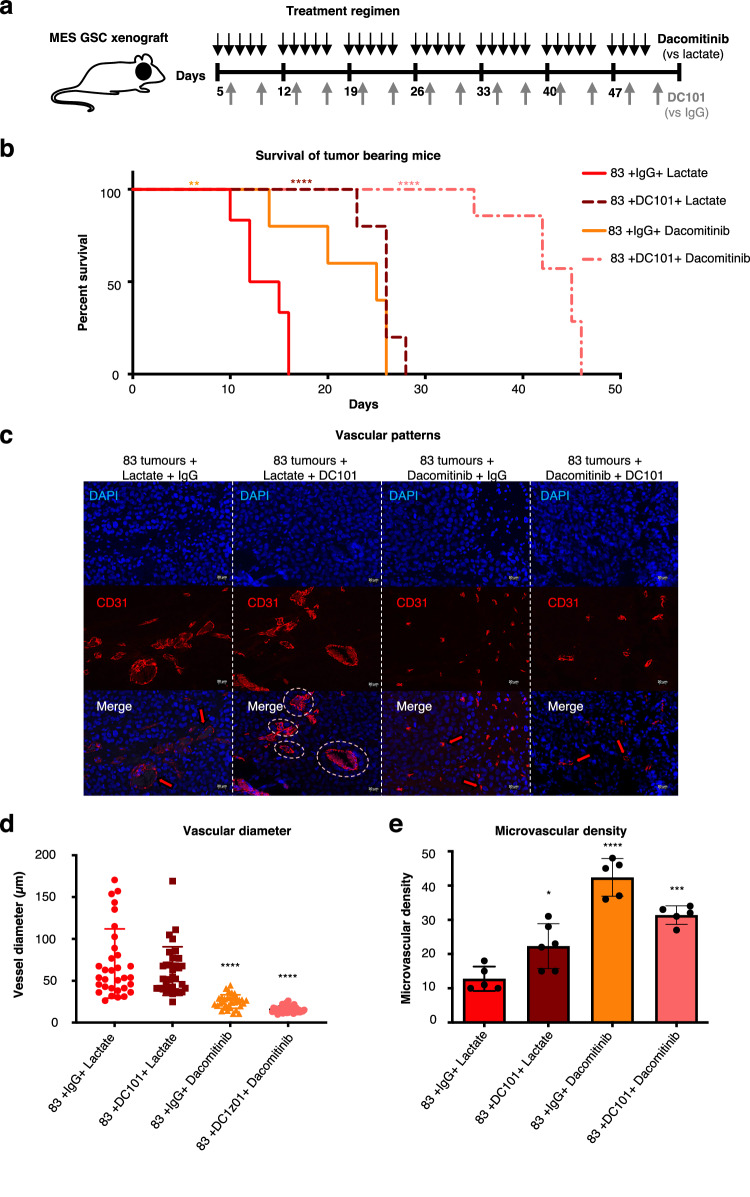
Targeting neovascularization in glioblastoma (GBM) is hampered by poor understanding of the underlying mechanisms and unclear linkages to tumour molecular landscapes. Here we report that different molecular subtypes of human glioma stem cells (GSC) trigger distinct endothelial responses involving either angiogenic or circumferential vascular growth (vasectasia). The latter process is selectively triggered by mesenchymal (but not proneural) GSCs and is mediated by a subset of extracellular vesicles (EVs) able to transfer EGFR/EGFRvIII transcript to endothelial cells. Inhibition of the expression and phosphorylation of EGFR in endothelial cells, either pharmacologically (Dacomitinib) or genetically (gene editing), abolishes their EV responses in vitro and disrupts vasectasia in vivo. Therapeutic inhibition of EGFR markedly extends anticancer effects of VEGF blockade in mice, coupled with abrogation of vasectasia and prolonged survival. Thus, vasectasia driven by intercellular transfer of oncogenic EGFR may represent a new therapeutic target in a subset of GBMs.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
- 39799/Canada Foundation for Innovation (Fondation canadienne pour l'innovation)
- PJT 183971/Gouvernement du Canada | Canadian Institutes of Health Research (Instituts de Recherche en Santé du Canada)
- N/A/Kidney Foundation of Canada (La Fondation canadienne du rein)
- N/A/U.S. Department of Defense (United States Department of Defense)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
