

Improving prime editing with an endogenous small RNA-binding protein
- PMID: 38570691
- PMCID: PMC11023932
- DOI: 10.1038/s41586-024-07259-6
Improving prime editing with an endogenous small RNA-binding protein
Abstract
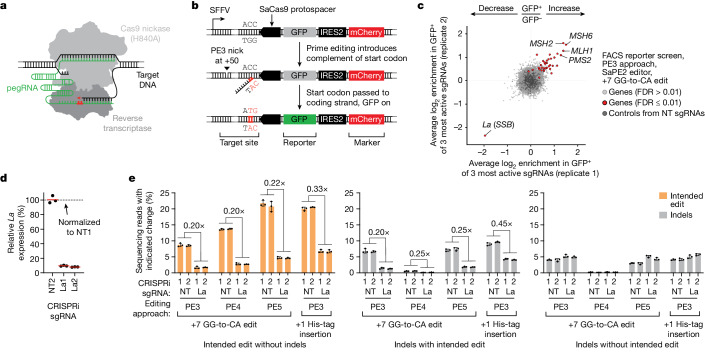
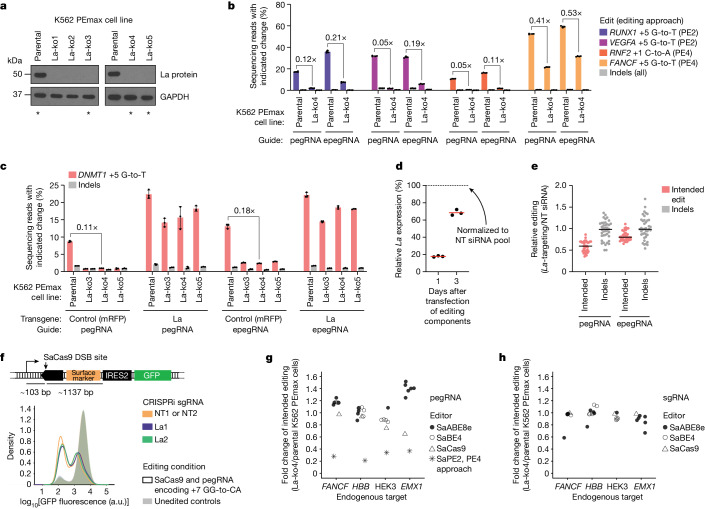
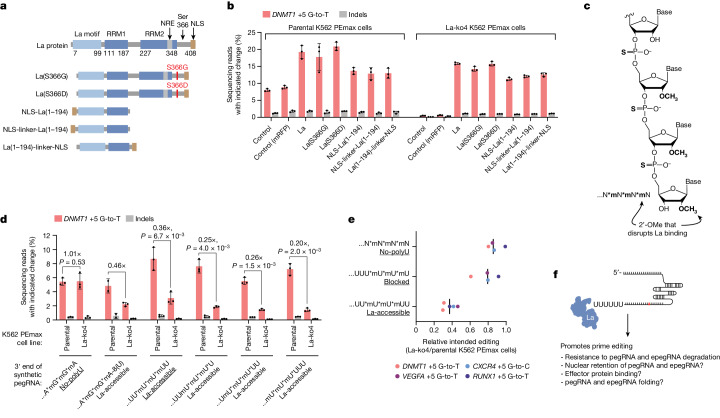
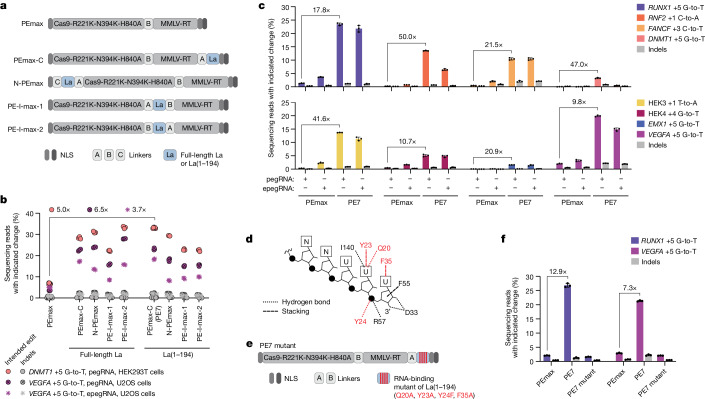
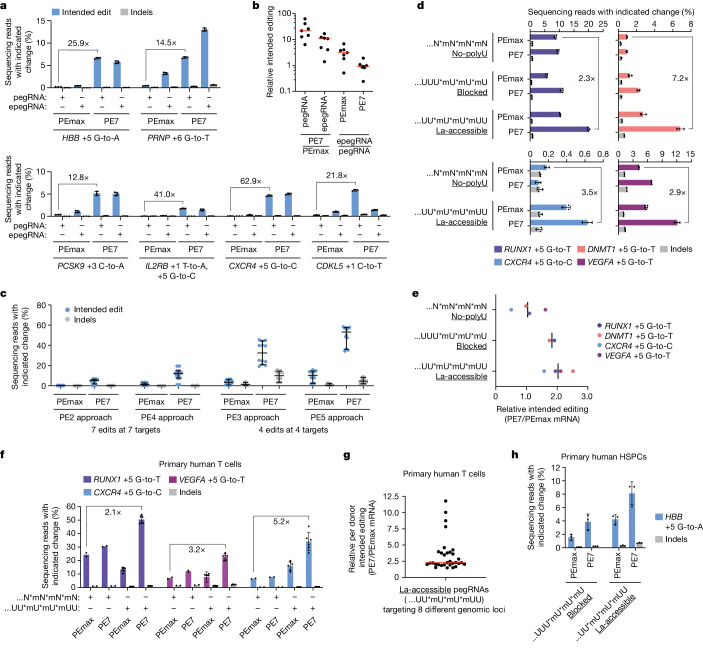
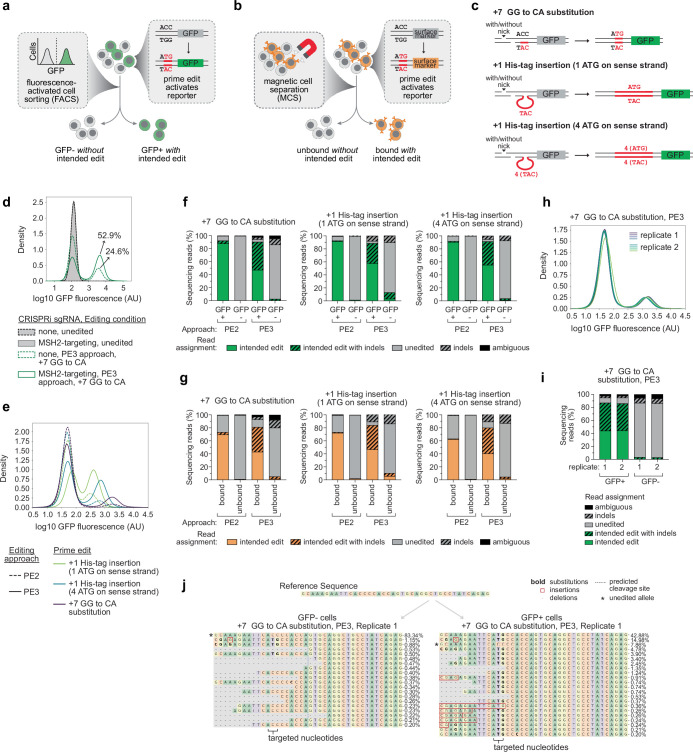
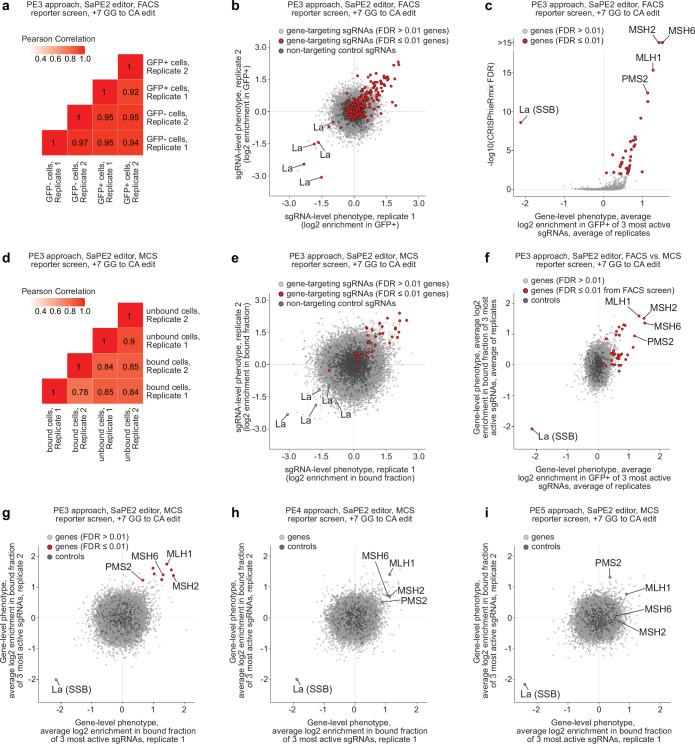
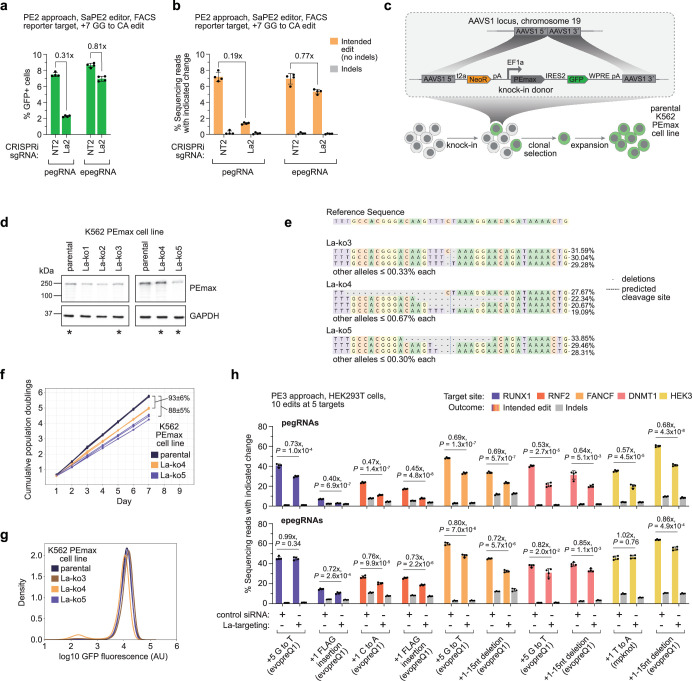
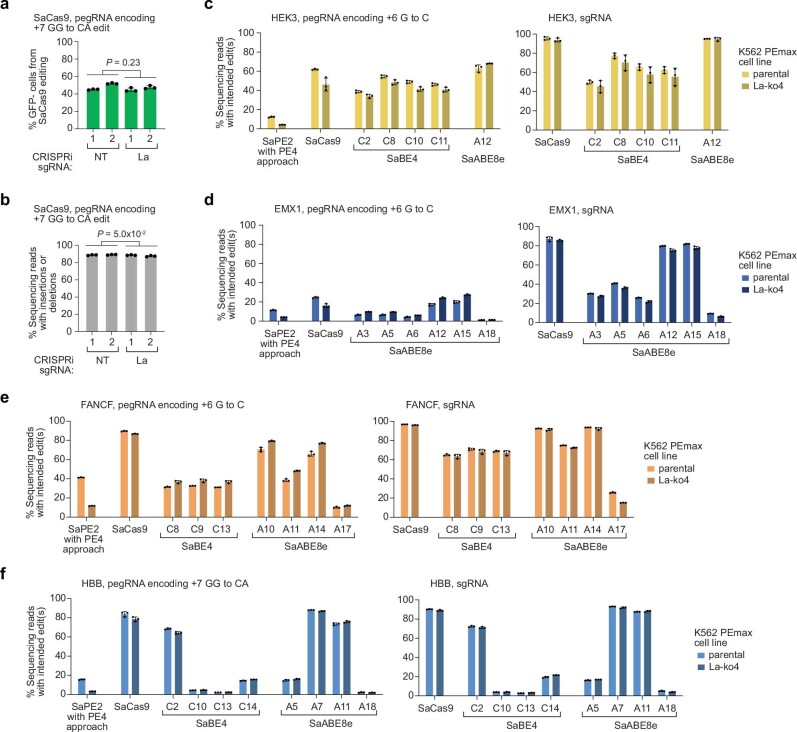
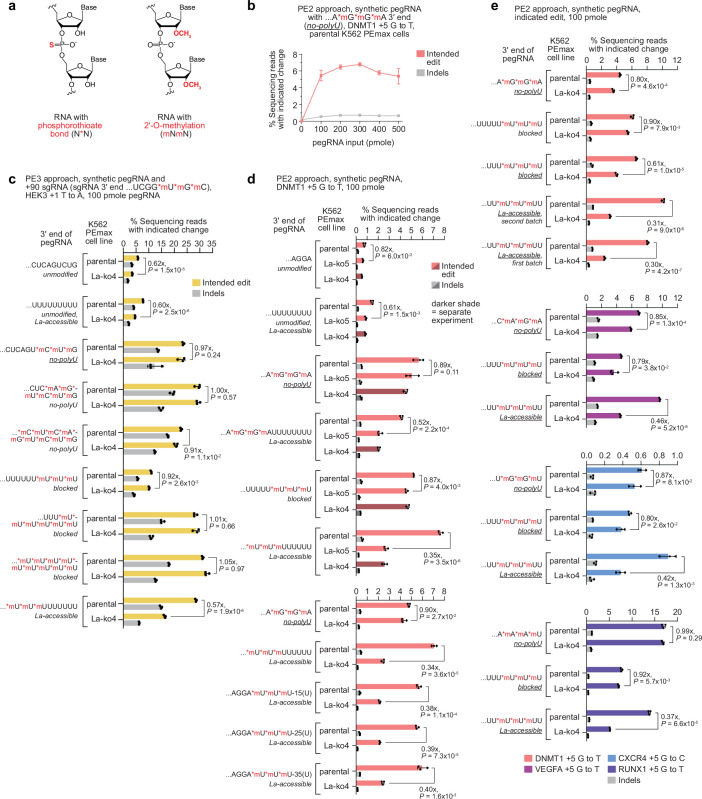
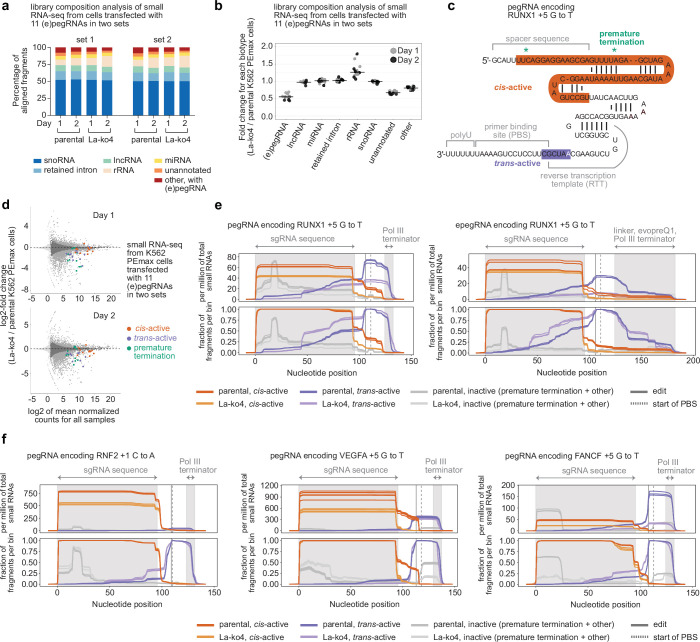
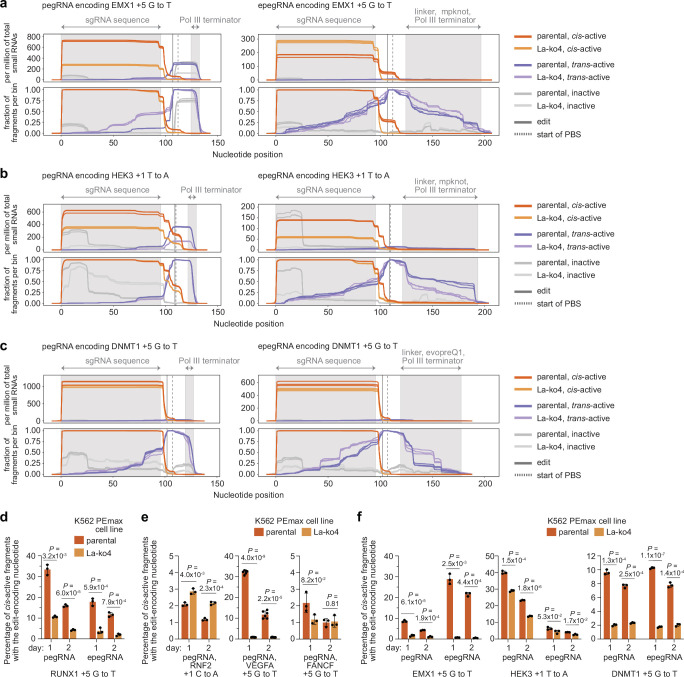
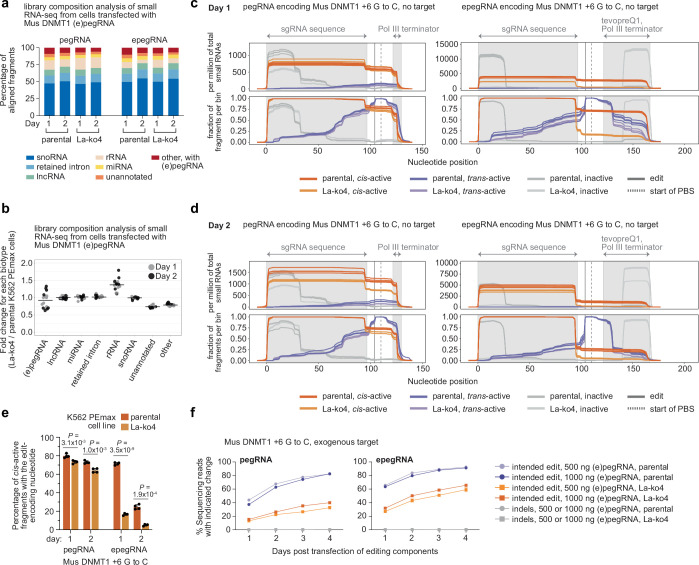
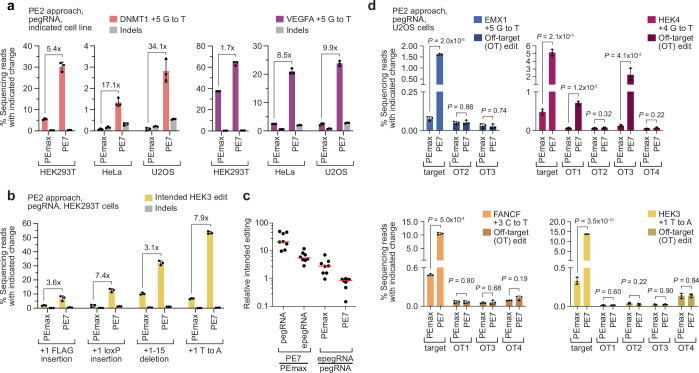
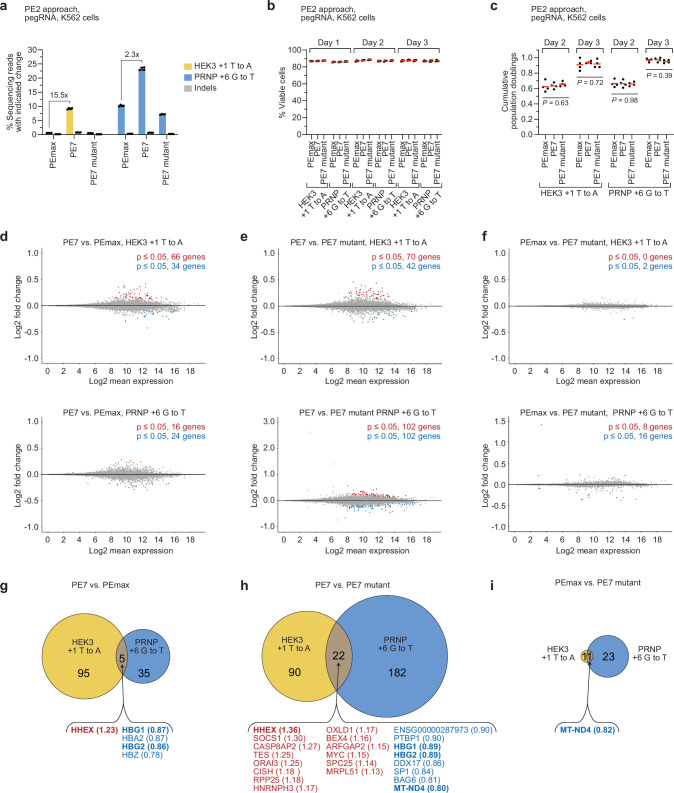
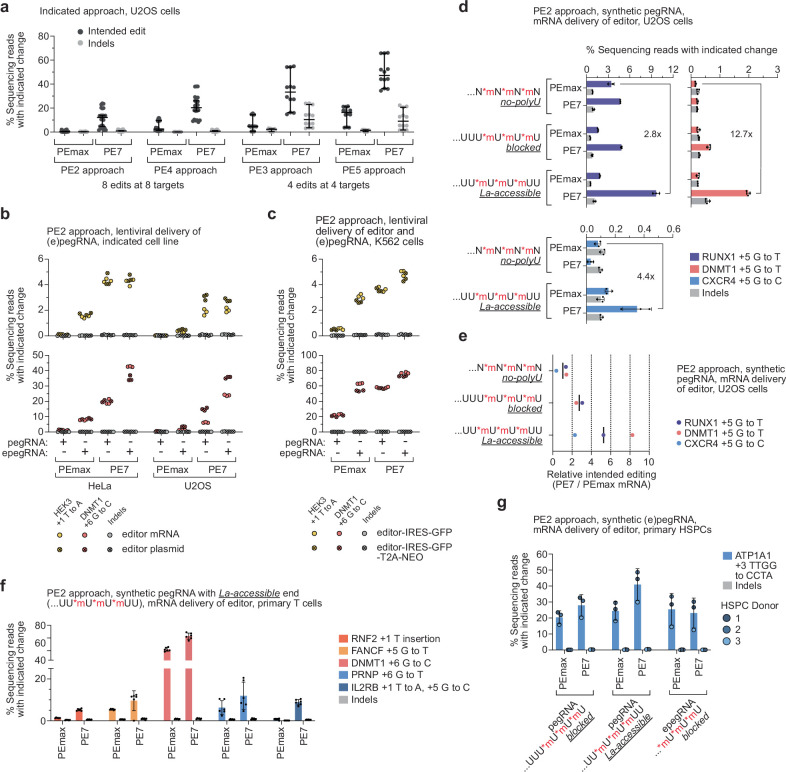
Prime editing enables the precise modification of genomes through reverse transcription of template sequences appended to the 3' ends of CRISPR-Cas guide RNAs1. To identify cellular determinants of prime editing, we developed scalable prime editing reporters and performed genome-scale CRISPR-interference screens. From these screens, a single factor emerged as the strongest mediator of prime editing: the small RNA-binding exonuclease protection factor La. Further investigation revealed that La promotes prime editing across approaches (PE2, PE3, PE4 and PE5), edit types (substitutions, insertions and deletions), endogenous loci and cell types but has no consistent effect on genome-editing approaches that rely on standard, unextended guide RNAs. Previous work has shown that La binds polyuridine tracts at the 3' ends of RNA polymerase III transcripts2. We found that La functionally interacts with the 3' ends of polyuridylated prime editing guide RNAs (pegRNAs). Guided by these results, we developed a prime editor protein (PE7) fused to the RNA-binding, N-terminal domain of La. This editor improved prime editing with expressed pegRNAs and engineered pegRNAs (epegRNAs), as well as with synthetic pegRNAs optimized for La binding. Together, our results provide key insights into how prime editing components interact with the cellular environment and suggest general strategies for stabilizing exogenous small RNAs therein.
© 2024. The Author(s).
Conflict of interest statement
B.A. is an advisory board member with options for Arbor Biotechnologies and Tessera Therapeutics. B.A. holds equity in Celsius Therapeutics. L.A.G has filed patents on CRISPR tools and CRISPR functional genomics and is a co-founder of Chroma Medicine. A.M. is a co-founder of Arsenal Biosciences, Site Tx, Spotlight Therapeutics, and Survey Genomics, serves on the boards of directors at Site Tx, Spotlight Therapeutics and Survey Genomics, is a member of the scientific advisory boards of Arsenal Biosciences, Site Tx, Spotlight Therapeutics, Survey Genomics, NewLimit, Amgen, Tenaya, and Lightcast, owns stock in Arsenal Biosciences, Site Tx, Spotlight Therapeutics, NewLimit, Survey Genomics, PACT Pharma, Tenaya, and Lightcast, and has received fees from Arsenal Biosciences, Spotlight Therapeutics, Site Tx NewLimit, Survey Genomics, Gilead, 23andMe, PACT Pharma, Juno Therapeutics, Tenaya, Lightcast, Trizell, Vertex, Merck, Amgen, Genentech, AlphaSights, Rupert Case Management, Bernstein, GLG, ClearView Healthcare Partners, and ALDA. A.M. is an investor in and informal advisor to Offline Ventures and a client of EPIQ. The Marson Laboratory has received research support from Juno Therapeutics, Epinomics, Sanofi, GlaxoSmithKline, Gilead, and Anthem. C.C.W. and R.S. are co-founders of Site Tx. J.Y. and B.A. have filed a patent application on aspects of this work through Princeton University, and B.A. has previously filed other patents on CRISPR-based technologies. The remaining authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
