

m6A-dependent upregulation of DDX21 by super-enhancer-driven IGF2BP2 and IGF2BP3 facilitates progression of acute myeloid leukaemia
- PMID: 38572589
- PMCID: PMC10993053
- DOI: 10.1002/ctm2.1628
m6A-dependent upregulation of DDX21 by super-enhancer-driven IGF2BP2 and IGF2BP3 facilitates progression of acute myeloid leukaemia
Abstract
Background: Acute myeloid leukaemia (AML) is a haematological malignancy with unfavourable prognosis. Despite the effectiveness of chemotherapy and targeted therapy, relapse or drug resistance remains a major threat to AML patients. N6-methyladenosine (m6A) RNA methylation and super-enhancers (SEs) are extensively involved in the leukaemogenesis of AML. However, the potential relationship between m6A and SEs in AML has not been elaborated.
Methods: Chromatin immunoprecipitation (ChIP) sequencing data from Gene Expression Omnibus (GEO) cohort were analysed to search SE-related genes. The mechanisms of m6 A-binding proteins IGF2BP2 and IGF2BP3 on DDX21 were explored via methylated RNA immunoprecipitation (MeRIP) assays, RNA immunoprecipitation (RIP) assays and luciferase reporter assays. Then we elucidated the roles of DDX21 in AML through functional assays in vitro and in vivo. Finally, co-immunoprecipitation (Co-IP) assays, RNA sequencing and ChIP assays were performed to investigate the downstream mechanisms of DDX21.
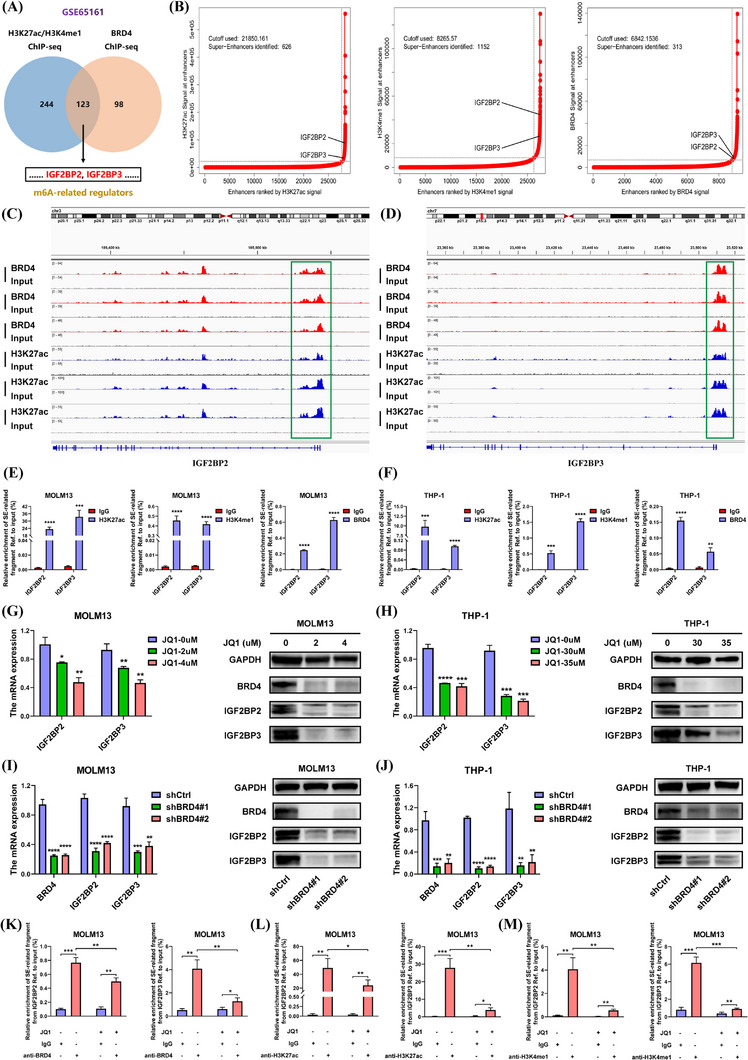
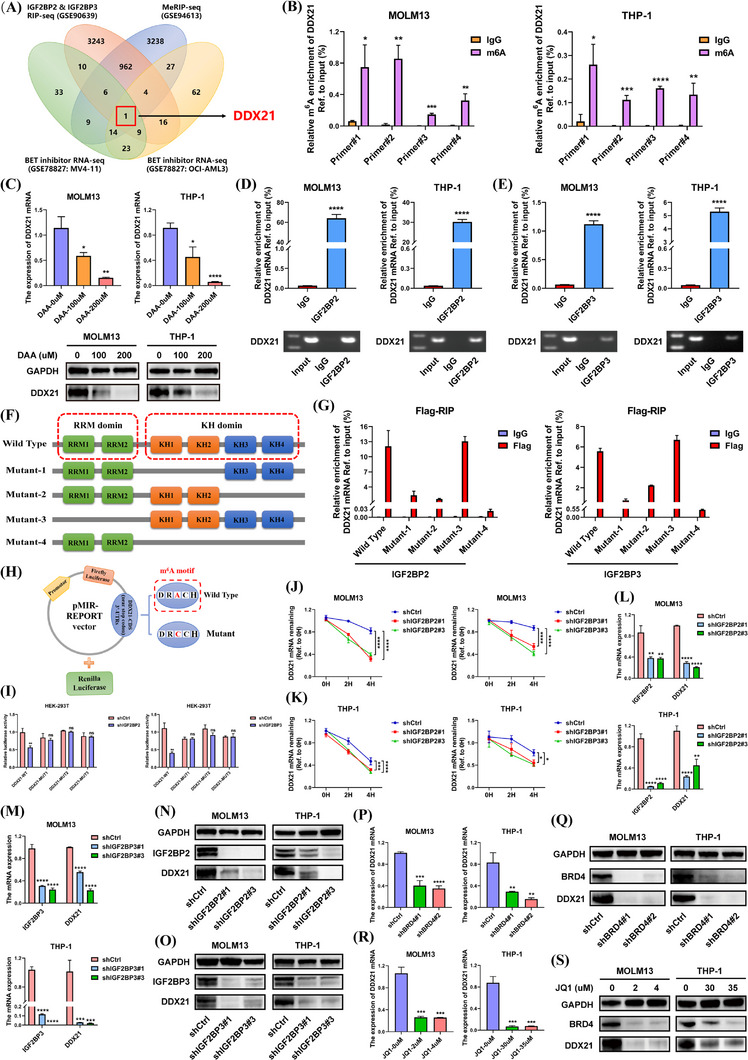
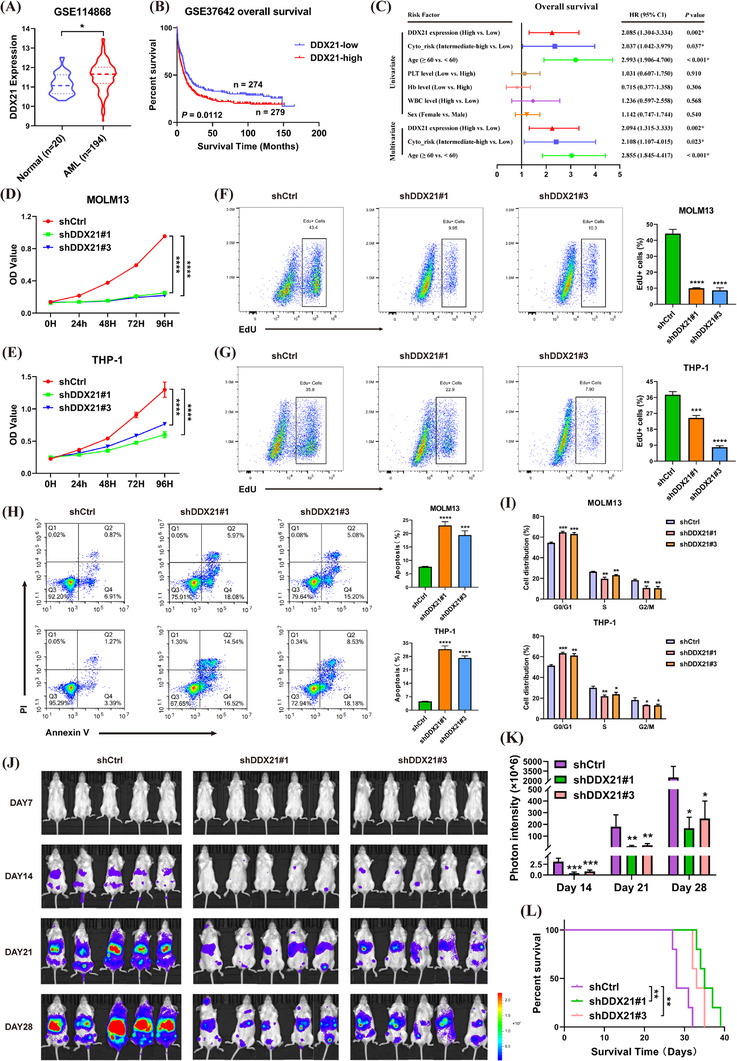
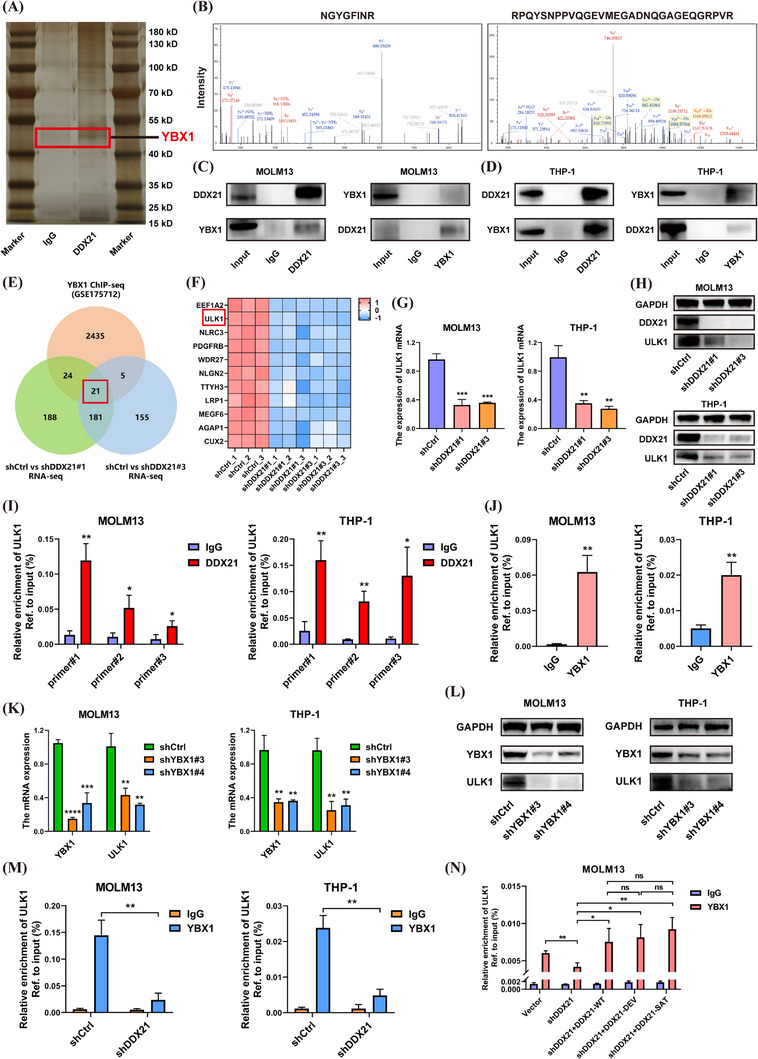
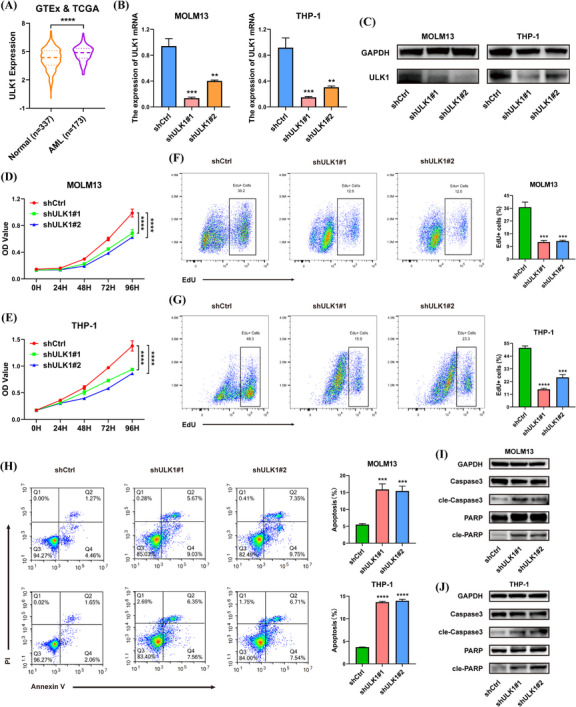
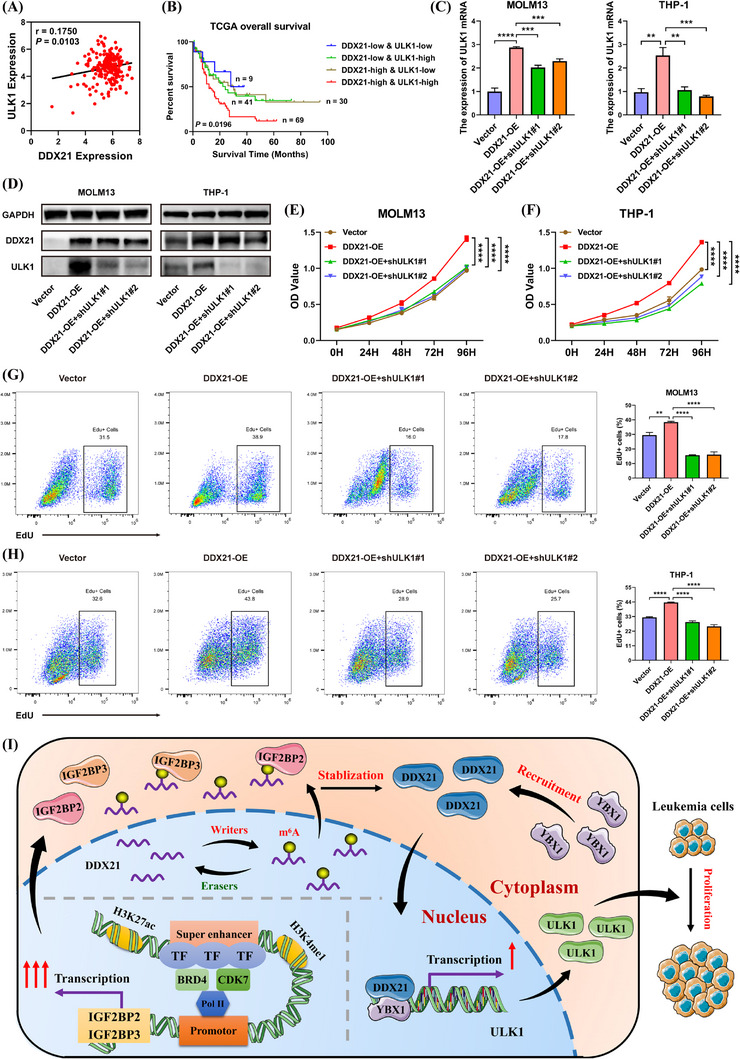
Results: We identified two SE-associated transcripts IGF2BP2 and IGF2BP3 in AML. High enrichment of H3K27ac, H3K4me1 and BRD4 was observed in IGF2BP2 and IGF2BP3, whose expression were driven by SE machinery. Then IGF2BP2 and IGF2BP3 enhanced the stability of DDX21 mRNA in an m6A-dependent manner. DDX21 was highly expressed in AML patients, which indicated a poor survival. Functionally, knockdown of DDX21 inhibited cell proliferation, promoted cell apoptosis and led to cell cycle arrest. Mechanistically, DDX21 recruited transcription factor YBX1 to cooperatively trigger ULK1 expression. Moreover, silencing of ULK1 could reverse the promoting effects of DDX21 overexpression in AML cells.
Conclusions: Dysregulation of SE-IGF2BP2/IGF2BP3-DDX21 axis facilitated the progression of AML. Our findings provide new insights into the link between SEs and m6A modification, elucidate the regulatory mechanisms of IGF2BP2 and IGF2BP3 on DDX21, and reveal the underlying roles of DDX21 in AML.
Keywords: DDX21; N6‐methyladenosine (m6A); acute myeloid leukaemia (AML); super‐enhancers (SEs).
© 2024 The Authors. Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
The authors declare they have no conflicts of interest.
Figures
Similar articles
-
KLF7 regulates super-enhancer-driven IGF2BP2 overexpression to promote the progression of head and neck squamous cell carcinoma.J Exp Clin Cancer Res. 2024 Mar 5;43(1):69. doi: 10.1186/s13046-024-02996-y. J Exp Clin Cancer Res. 2024. PMID: 38443991 Free PMC article.
-
N6-methyladenosine-modified SENP1, identified by IGF2BP3, is a novel molecular marker in acute myeloid leukemia and aggravates progression by activating AKT signal via de-SUMOylating HDAC2.Mol Cancer. 2024 May 31;23(1):116. doi: 10.1186/s12943-024-02013-y. Mol Cancer. 2024. PMID: 38822351 Free PMC article.
-
LINC00987 knockdown inhibits the progression of acute myeloid leukemia by suppressing IGF2BP2-mediated PA2G4 expression.Anticancer Drugs. 2022 Jan 1;33(1):e207-e217. doi: 10.1097/CAD.0000000000001188. Anticancer Drugs. 2022. PMID: 34407052
-
Current understanding of the role of DDX21 in orchestrating gene expression in health and diseases.Life Sci. 2024 Jul 15;349:122716. doi: 10.1016/j.lfs.2024.122716. Epub 2024 May 16. Life Sci. 2024. PMID: 38762067 Review.
-
STAT3 Facilitates Super Enhancer Formation to Promote Fibroblast-To-Myofibroblast Differentiation by the Analysis of ATAC-Seq, RNA-Seq and ChIP-Seq.J Cell Mol Med. 2025 Jun;29(11):e70639. doi: 10.1111/jcmm.70639. J Cell Mol Med. 2025. PMID: 40464702 Free PMC article. Review.
Cited by
-
DNA methylation in monozygotic twins discordant for acute lymphoblastic leukemia: a case report and systematic review.Clin Epigenetics. 2025 Jun 6;17(1):94. doi: 10.1186/s13148-025-01906-z. Clin Epigenetics. 2025. PMID: 40481591 Free PMC article. Review.
-
The Emerging Role of IGF2BP2 in Cancer Therapy Resistance: From Molecular Mechanism to Future Potential.Int J Mol Sci. 2024 Nov 12;25(22):12150. doi: 10.3390/ijms252212150. Int J Mol Sci. 2024. PMID: 39596216 Free PMC article. Review.
-
Multifaceted roles of insulin‑like growth factor 2 mRNA binding protein 2 in human cancer (Review).Mol Med Rep. 2025 Mar;31(3):75. doi: 10.3892/mmr.2025.13441. Epub 2025 Jan 31. Mol Med Rep. 2025. PMID: 39886962 Free PMC article. Review.
-
DDX21 at the Nexus of RNA Metabolism, Cancer Oncogenesis, and Host-Virus Crosstalk: Decoding Its Biomarker Potential and Therapeutic Implications.Int J Mol Sci. 2024 Dec 19;25(24):13581. doi: 10.3390/ijms252413581. Int J Mol Sci. 2024. PMID: 39769343 Free PMC article. Review.
-
DDX39B K63-linked ubiquitination mediated by TRIM28 promotes NSCLC metastasis by enhancing ECAD lysosomal degradation.Signal Transduct Target Ther. 2025 Jul 16;10(1):221. doi: 10.1038/s41392-025-02305-9. Signal Transduct Target Ther. 2025. PMID: 40664668 Free PMC article.
References
-
- Lowenberg B, Downing J, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051‐1062. - PubMed
-
- Juliusson G, Antunovic P, Derolf A, et al. Age and acute myeloid leukemia: real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood. 2009;113:4179‐4187. - PubMed
-
- Döhner H, Wei A, Löwenberg B. Towards precision medicine for AML. Nat Rev Clin Oncol. 2021;18:577‐590. - PubMed
-
- Shallis RM, Wang R, Davidoff A, et al. Epidemiology of acute myeloid leukemia: recent progress and enduring challenges. Blood Rev. 2019;36:70‐87. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous