

Genetics and Genomics of Pulmonary Fibrosis: Charting the Molecular Landscape and Shaping Precision Medicine
- PMID: 38573068
- PMCID: PMC11351799
- DOI: 10.1164/rccm.202401-0238SO
Genetics and Genomics of Pulmonary Fibrosis: Charting the Molecular Landscape and Shaping Precision Medicine
Abstract
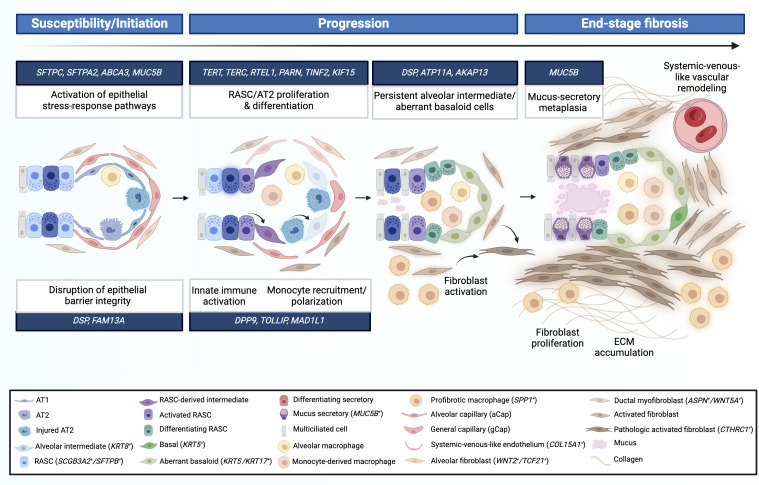
Recent genetic and genomic advancements have elucidated the complex etiology of idiopathic pulmonary fibrosis (IPF) and other progressive fibrotic interstitial lung diseases (ILDs), emphasizing the contribution of heritable factors. This state-of-the-art review synthesizes evidence on significant genetic contributors to pulmonary fibrosis (PF), including rare genetic variants and common SNPs. The MUC5B promoter variant is unusual, a common SNP that markedly elevates the risk of early and established PF. We address the utility of genetic variation in enhancing understanding of disease pathogenesis and clinical phenotypes, improving disease definitions, and informing prognosis and treatment response. Critical research gaps are highlighted, particularly the underrepresentation of non-European ancestries in PF genetic studies and the exploration of PF phenotypes beyond usual interstitial pneumonia/IPF. We discuss the role of telomere length, often critically short in PF, and its link to progression and mortality, underscoring the genetic complexity involving telomere biology genes (TERT, TERC) and others like SFTPC and MUC5B. In addition, we address the potential of gene-by-environment interactions to modulate disease manifestation, advocating for precision medicine in PF. Insights from gene expression profiling studies and multiomic analyses highlight the promise for understanding disease pathogenesis and offer new approaches to clinical care, therapeutic drug development, and biomarker discovery. Finally, we discuss the ethical, legal, and social implications of genomic research and therapies in PF, stressing the need for sound practices and informed clinical genetic discussions. Looking forward, we advocate for comprehensive genetic testing panels and polygenic risk scores to improve the management of PF and related ILDs across diverse populations.
Keywords: MUC5B promoter variant; genetic variants; interstitial lung diseases; precision medicine; pulmonary fibrosis.
Figures



Similar articles
-
Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways.Physiol Rev. 2016 Oct;96(4):1567-91. doi: 10.1152/physrev.00004.2016. Physiol Rev. 2016. PMID: 27630174 Free PMC article. Review.
-
Long-Term Air Pollution Exposure and Severity of Idiopathic Pulmonary Fibrosis: Data from the Idiopathic Pulmonary Fibrosis Prospective Outcomes (IPF-PRO) Registry.Ann Am Thorac Soc. 2025 Mar;22(3):378-386. doi: 10.1513/AnnalsATS.202404-382OC. Ann Am Thorac Soc. 2025. PMID: 39531618
-
Current Understanding of Pulmonary Fibrosis: Pathogenesis, Diagnosis, and Therapeutic Approaches.Can Respir J. 2025 Jul 15;2025:3183241. doi: 10.1155/carj/3183241. eCollection 2025. Can Respir J. 2025. PMID: 40697404 Free PMC article. Review.
-
Epithelial Endoplasmic Reticulum Stress Enhances the Risk of Muc5b-associated Lung Fibrosis.Am J Respir Cell Mol Biol. 2023 Jan;68(1):62-74. doi: 10.1165/rcmb.2022-0252OC. Am J Respir Cell Mol Biol. 2023. PMID: 36108173 Free PMC article.
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
Cited by
-
Identification of core genes in the extracellular matrix and the regulatory mechanisms of the immune microenvironment in idiopathic pulmonary fibrosis using WGCNA and machine learning methods.PLoS One. 2025 Aug 26;20(8):e0330725. doi: 10.1371/journal.pone.0330725. eCollection 2025. PLoS One. 2025. PMID: 40857336 Free PMC article.
-
[Research Progress of Anti-lung Cancer Drug-related Interstitial Lung Disease].Zhongguo Fei Ai Za Zhi. 2025 Apr 20;28(4):309-318. doi: 10.3779/j.issn.1009-3419.2025.106.11. Zhongguo Fei Ai Za Zhi. 2025. PMID: 40404479 Free PMC article. Review. Chinese.
-
Biomarker-defined endotypes of pulmonary fibrosis.Lancet Respir Med. 2024 Sep;12(9):657-659. doi: 10.1016/S2213-2600(24)00169-3. Epub 2024 Jul 15. Lancet Respir Med. 2024. PMID: 39025090 Free PMC article. No abstract available.
-
Genetic evidence reveals a causal relationship between rheumatoid arthritis and interstitial lung disease.Front Genet. 2024 May 14;15:1395315. doi: 10.3389/fgene.2024.1395315. eCollection 2024. Front Genet. 2024. PMID: 38808332 Free PMC article.
-
Assessment of tumor biomarkers for prognosis in interstitial lung disease associated with connective tissue disease: a prospective study.J Thorac Dis. 2024 Nov 30;16(11):7383-7396. doi: 10.21037/jtd-24-922. Epub 2024 Nov 21. J Thorac Dis. 2024. PMID: 39678894 Free PMC article.
References
-
- Rhodes CJ, Batai K, Bleda M, Haimel M, Southgate L, Germain M, et al. UK NIHR BioResource Rare Diseases Consortium UK PAH Cohort Study Consortium; US PAH Biobank Consortium. Genetic determinants of risk in pulmonary arterial hypertension: international genome-wide association studies and meta-analysis. Lancet Respir Med . 2019;7:227–238. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K23 HL146942/HL/NHLBI NIH HHS/United States
- UH3 HL151865/HL/NHLBI NIH HHS/United States
- R01HL153246/HL/NHLBI NIH HHS/United States
- I01 BX006121/BX/BLRD VA/United States
- R01 HL169166/HL/NHLBI NIH HHS/United States
- K23HL146942/HL/NHLBI NIH HHS/United States
- I01 BX002378/BX/BLRD VA/United States
- UH3 HL145266/HL/NHLBI NIH HHS/United States
- P01 HL092870/HL/NHLBI NIH HHS/United States
- R01 HL158668/HL/NHLBI NIH HHS/United States
- R01 HL130974/HL/NHLBI NIH HHS/United States
- R01HG011886/HL/NHLBI NIH HHS/United States
- R01 HL145372/HL/NHLBI NIH HHS/United States
- R01 HL130796/HL/NHLBI NIH HHS/United States
- R01 HL111024/HL/NHLBI NIH HHS/United States
- R01HL145372/HL/NHLBI NIH HHS/United States
- R01 HL135142/HL/NHLBI NIH HHS/United States
- I01 BX005295/BX/BLRD VA/United States
- P01 HL162607/HL/NHLBI NIH HHS/United States
- K23 HL150301/HL/NHLBI NIH HHS/United States
- R01 HL149836/HL/NHLBI NIH HHS/United States
- R01 HG011886/HG/NHGRI NIH HHS/United States
- R01 HL153246/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
