

Adaptive functions of structural variants in human brain development
- PMID: 38579006
- PMCID: PMC11092980
- DOI: 10.1126/sciadv.adl4600
Adaptive functions of structural variants in human brain development
Abstract
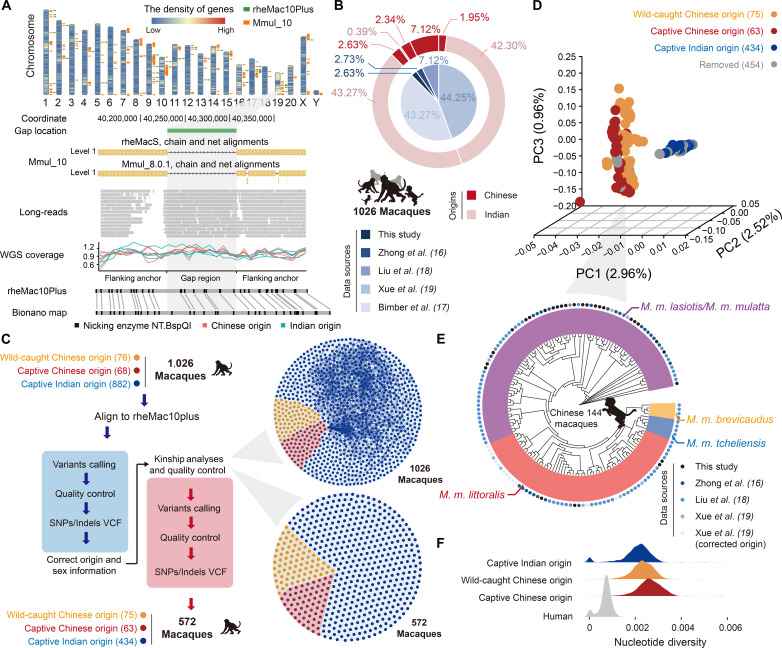
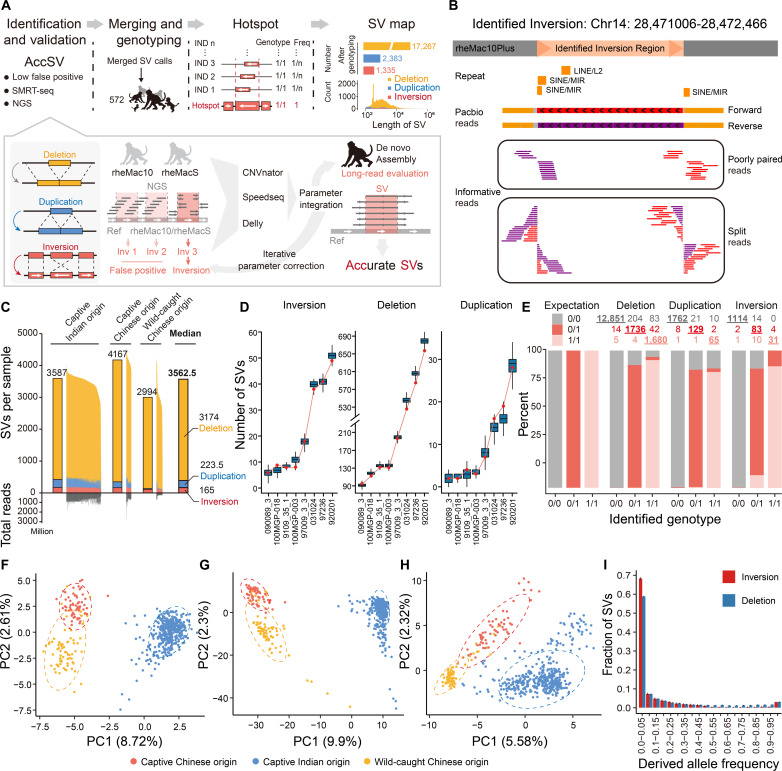
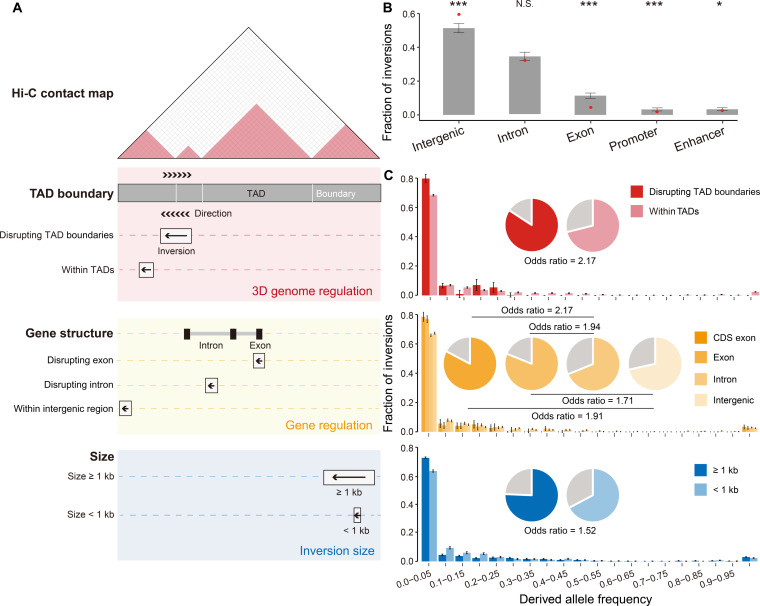
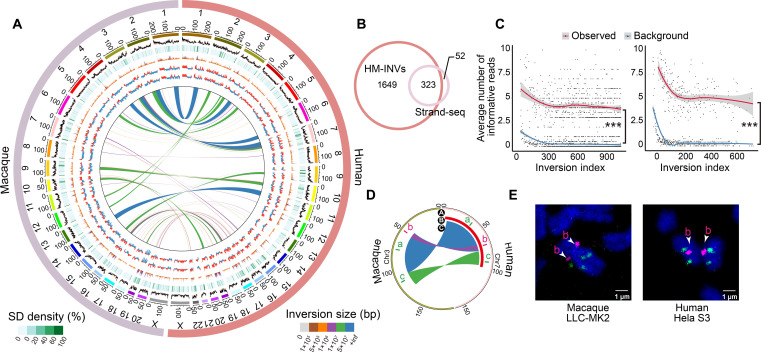
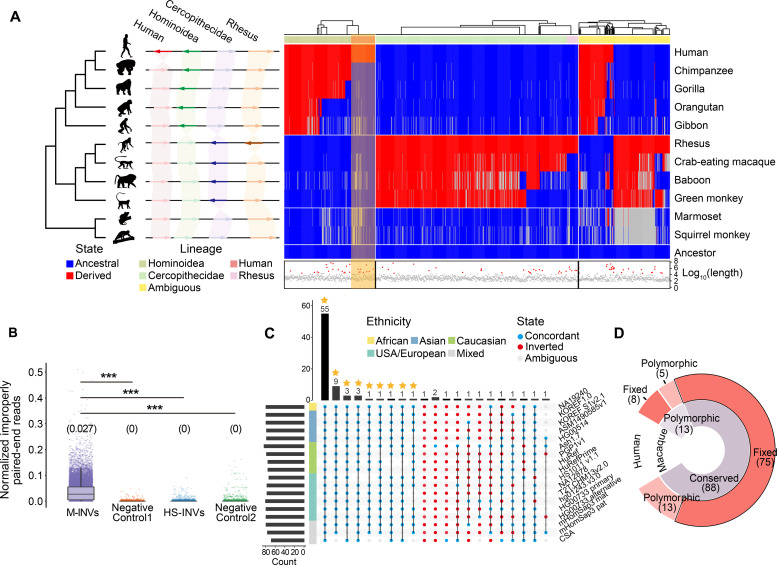
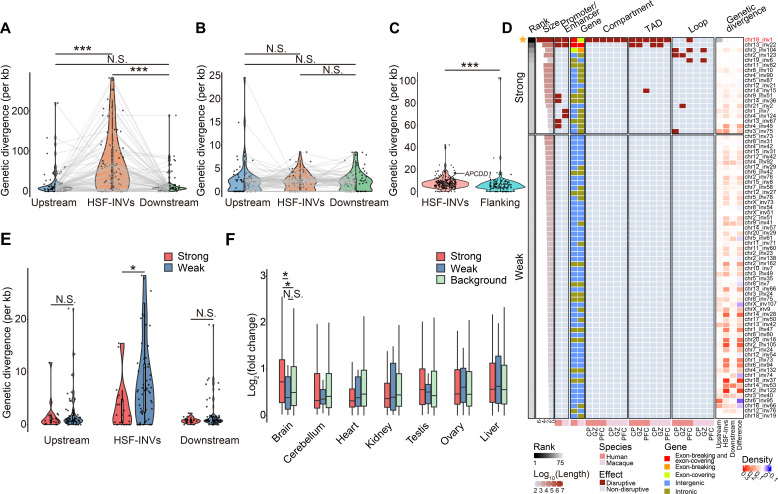
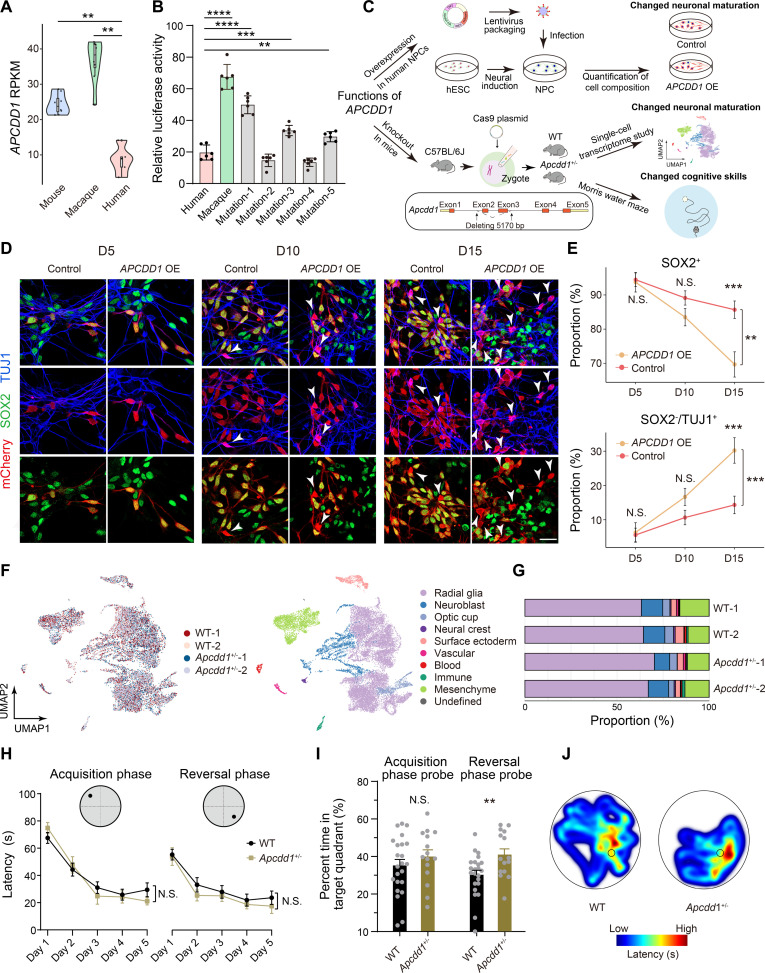
Quantifying the structural variants (SVs) in nonhuman primates could provide a niche to clarify the genetic backgrounds underlying human-specific traits, but such resource is largely lacking. Here, we report an accurate SV map in a population of 562 rhesus macaques, verified by in-house benchmarks of eight macaque genomes with long-read sequencing and another one with genome assembly. This map indicates stronger selective constrains on inversions at regulatory regions, suggesting a strategy for prioritizing them with the most important functions. Accordingly, we identified 75 human-specific inversions and prioritized them. The top-ranked inversions have substantially shaped the human transcriptome, through their dual effects of reconfiguring the ancestral genomic architecture and introducing regional mutation hotspots at the inverted regions. As a proof of concept, we linked APCDD1, located on one of these inversions and down-regulated specifically in humans, to neuronal maturation and cognitive ability. We thus highlight inversions in shaping the human uniqueness in brain development.
Figures
Similar articles
-
Single-cell strand sequencing of a macaque genome reveals multiple nested inversions and breakpoint reuse during primate evolution.Genome Res. 2020 Nov;30(11):1680-1693. doi: 10.1101/gr.265322.120. Epub 2020 Oct 22. Genome Res. 2020. PMID: 33093070 Free PMC article.
-
Genomic structural variation: A complex but important driver of human evolution.Am J Biol Anthropol. 2023 Aug;181 Suppl 76(Suppl 76):118-144. doi: 10.1002/ajpa.24713. Epub 2023 Feb 16. Am J Biol Anthropol. 2023. PMID: 36794631 Free PMC article. Review.
-
Unraveling the effect of genomic structural changes in the rhesus macaque - implications for the adaptive role of inversions.BMC Genomics. 2014 Jun 26;15(1):530. doi: 10.1186/1471-2164-15-530. BMC Genomics. 2014. PMID: 24969235 Free PMC article.
-
The population genomics of rhesus macaques (Macaca mulatta) based on whole-genome sequences.Genome Res. 2016 Dec;26(12):1651-1662. doi: 10.1101/gr.204255.116. Epub 2016 Oct 17. Genome Res. 2016. PMID: 27934697 Free PMC article.
-
Geographic distribution and adaptive significance of genomic structural variants: an anthropological genetics perspective.Hum Biol. 2014 Fall;86(4):260-75. doi: 10.13110/humanbiology.86.4.0260. Hum Biol. 2014. PMID: 25959693 Review.
Cited by
-
Deciphering the role of structural variation in human evolution: a functional perspective.Curr Opin Genet Dev. 2024 Oct;88:102240. doi: 10.1016/j.gde.2024.102240. Epub 2024 Aug 8. Curr Opin Genet Dev. 2024. PMID: 39121701 Free PMC article. Review.
-
VCF2Dis: an ultra-fast and efficient tool to calculate pairwise genetic distance and construct population phylogeny from VCF files.Gigascience. 2025 Jan 6;14:giaf032. doi: 10.1093/gigascience/giaf032. Gigascience. 2025. PMID: 40184433 Free PMC article.
-
Human-specific gene expansions contribute to brain evolution.Cell. 2025 Jul 18:S0092-8674(25)00739-1. doi: 10.1016/j.cell.2025.06.037. Online ahead of print. Cell. 2025. PMID: 40695280
-
Human-specific gene expansions contribute to brain evolution.bioRxiv [Preprint]. 2025 Jun 25:2024.09.26.615256. doi: 10.1101/2024.09.26.615256. bioRxiv. 2025. Update in: Cell. 2025 Jul 18:S0092-8674(25)00739-1. doi: 10.1016/j.cell.2025.06.037. PMID: 39386494 Free PMC article. Updated. Preprint.
-
Long-read sequencing of hundreds of diverse brains provides insight into the impact of structural variation on gene expression and DNA methylation.bioRxiv [Preprint]. 2024 Dec 17:2024.12.16.628723. doi: 10.1101/2024.12.16.628723. bioRxiv. 2024. PMID: 39764002 Free PMC article. Preprint.
References
-
- Abel H. J., Larson D. E., Regier A. A., Chiang C., Das I., Kanchi K. L., Layer R. M., Neale B. M., Salerno W. J., Reeves C., Buyske S.; NHGRI Centers for Common Disease Genomics, Matise T. C., Muzny D. M., Zody M. C., Lander E. S., Dutcher S. K., Stitziel N. O., Hall I. M., Mapping and characterization of structural variation in 17,795 human genomes. Nature 583, 83–89 (2020). - PMC - PubMed
-
- Maggiolini F. A. M., Sanders A. D., Shew C. J., Sulovari A., Mao Y., Puig M., Catacchio C. R., Dellino M., Palmisano D., Mercuri L., Bitonto M., Porubský D., Cáceres M., Eichler E. E., Ventura M., Dennis M. Y., Korbel J. O., Antonacci F., Single-cell strand sequencing of a macaque genome reveals multiple nested inversions and breakpoint reuse during primate evolution. Genome Res. 30, 1680–1693 (2020). - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
