

Loss of the stress sensor GADD45A promotes stem cell activity and ferroptosis resistance in LGR4/HOXA9-dependent AML
- PMID: 38579286
- PMCID: PMC11251412
- DOI: 10.1182/blood.2024024072
Loss of the stress sensor GADD45A promotes stem cell activity and ferroptosis resistance in LGR4/HOXA9-dependent AML
Abstract
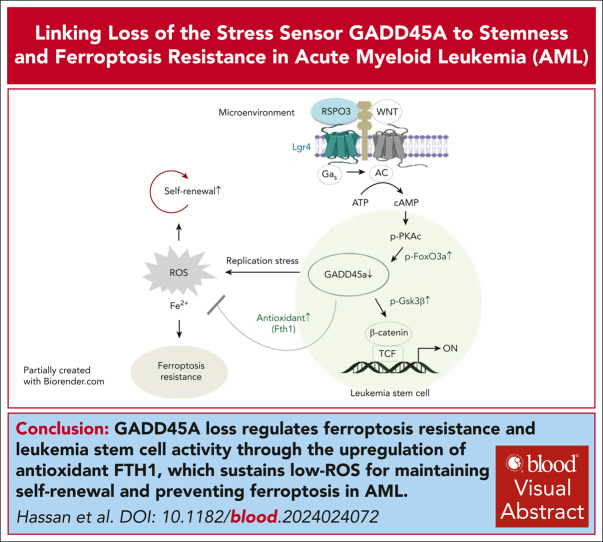
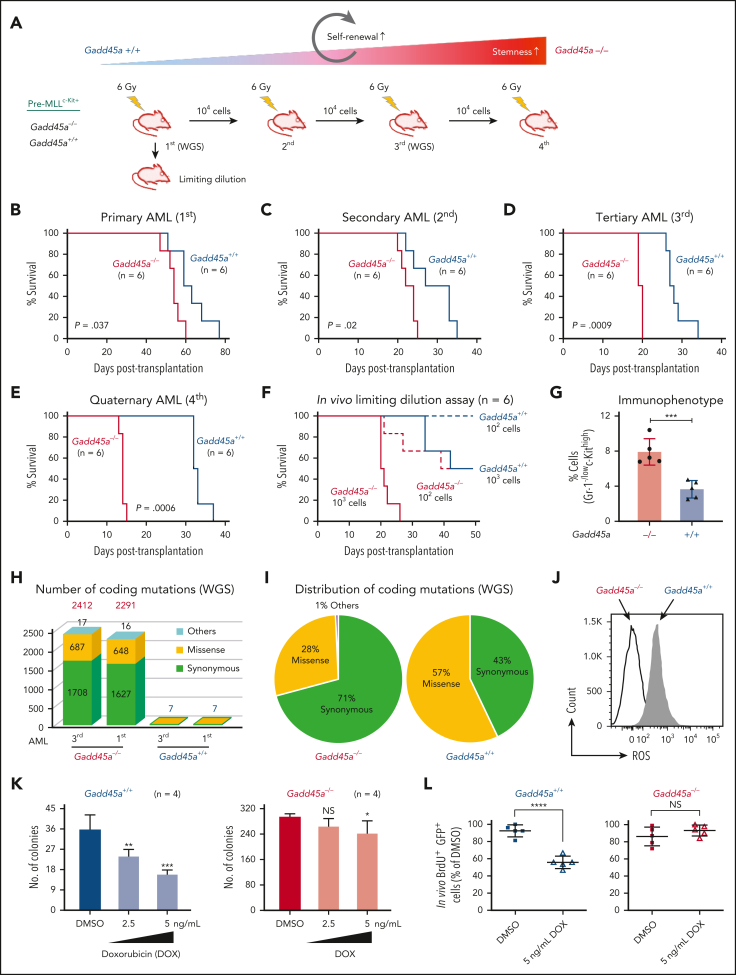
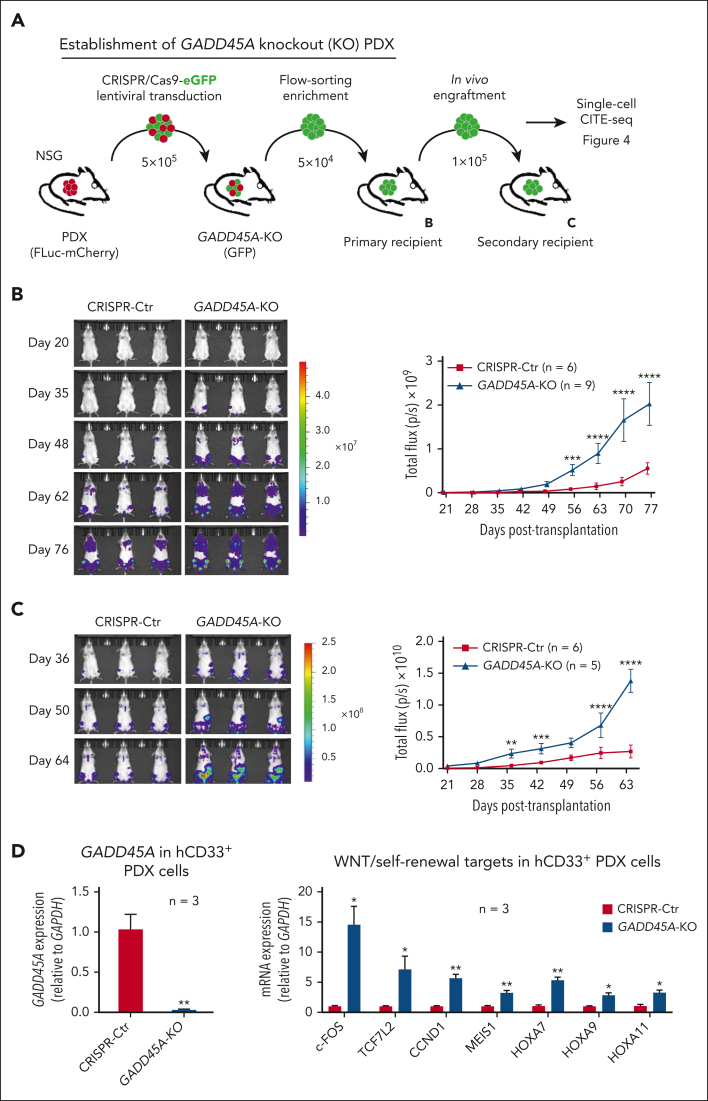
The overall prognosis of acute myeloid leukemia (AML) remains dismal, largely because of the inability of current therapies to kill leukemia stem cells (LSCs) with intrinsic resistance. Loss of the stress sensor growth arrest and DNA damage-inducible 45 alpha (GADD45A) is implicated in poor clinical outcomes, but its role in LSCs and AML pathogenesis is unknown. Here, we define GADD45A as a key downstream target of G protein-coupled receptor (LGR)4 pathway and discover a regulatory role for GADD45A loss in promoting leukemia-initiating activity and oxidative resistance in LGR4/HOXA9-dependent AML, a poor prognosis subset of leukemia. Knockout of GADD45A enhances AML progression in murine and patient-derived xenograft (PDX) mouse models. Deletion of GADD45A induces substantial mutations, increases LSC self-renewal and stemness in vivo, and reduces levels of reactive oxygen species (ROS), accompanied by a decreased response to ROS-associated genotoxic agents (eg, ferroptosis inducer RSL3) and acquisition of an increasingly aggressive phenotype on serial transplantation in mice. Our single-cell cellular indexing of transcriptomes and epitopes by sequencing analysis on patient-derived LSCs in PDX mice and subsequent functional studies in murine LSCs and primary AML patient cells show that loss of GADD45A is associated with resistance to ferroptosis (an iron-dependent oxidative cell death caused by ROS accumulation) through aberrant activation of antioxidant pathways related to iron and ROS detoxification, such as FTH1 and PRDX1, upregulation of which correlates with unfavorable outcomes in patients with AML. These results reveal a therapy resistance mechanism contributing to poor prognosis and support a role for GADD45A loss as a critical step for leukemia-initiating activity and as a target to overcome resistance in aggressive leukemia.
© 2024 American Society of Hematology. Published by Elsevier Inc. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures





Comment in
-
GADD45A: a key tumor suppressor in AML subtypes.Blood. 2024 Jul 4;144(1):6-7. doi: 10.1182/blood.2024024685. Blood. 2024. PMID: 38963670 No abstract available.
References
-
- Krivtsov AV, Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442(7104):818–822. - PubMed
-
- Stavropoulou V, Kaspar S, Brault L, et al. MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell. 2016;30(1):43–58. - PubMed
-
- Salik B, Yi H, Hassan N, et al. Targeting RSPO3-LGR4 signaling for leukemia stem cell eradication in acute myeloid leukemia. Cancer Cell. 2020;38(2):263–278.e6. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
