

Real-life experience with inotersen at CEPARM, Hospital Universitário Clementino Fraga Filho, Universidade Federal do Rio de Janeiro
- PMID: 38579737
- PMCID: PMC10997406
- DOI: 10.1055/s-0044-1781463
Real-life experience with inotersen at CEPARM, Hospital Universitário Clementino Fraga Filho, Universidade Federal do Rio de Janeiro
Abstract
Background: Hereditary transthyretin amyloidosis (ATTRv) is an inherited, progressive, and fatal disease still largely underdiagnosed. Mutations in the transthyretin (TTR) gene cause the TTR protein to destabilize, misfold, aggregate, and deposit in body tissues, which makes ATTRv a disease with heterogeneous clinical phenotype.
Objective: To describe the long-term efficacy and safety of inotersen therapy in patients with ATTRv peripheral neuropathy (ATTRv-PN).
Methods: Patients who completed the NEURO-TTR pivotal study and the NEURO-TTR OLE open-label extension study migrated to the present study and were followed-up for at least 18 more months to an average of 67 months and up to 76 months since day 1 of the inotersen therapy (D1-first dose of inotersen). Disease progression was evaluated by standard measures.
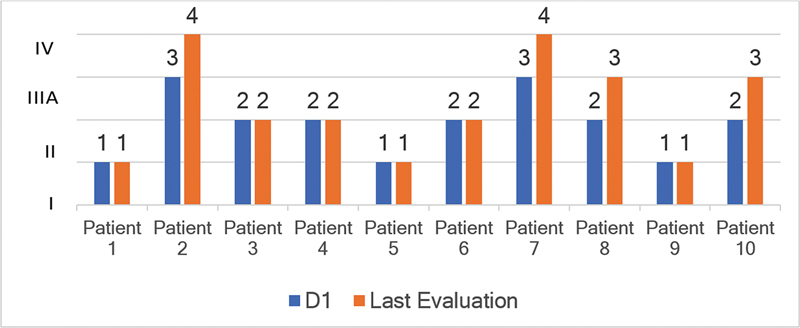
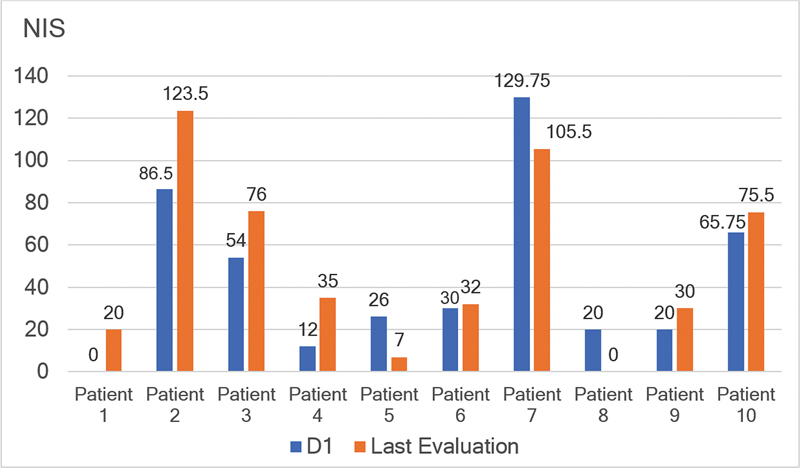
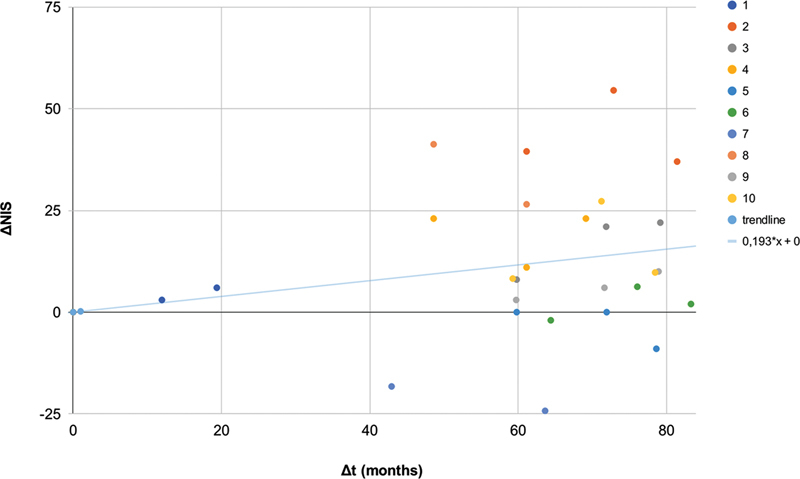
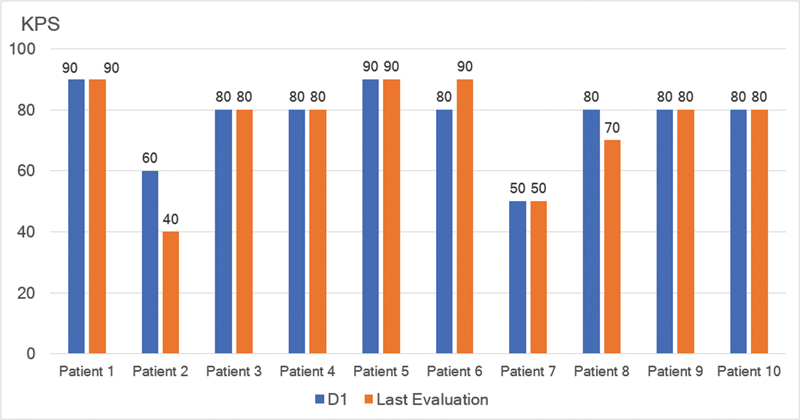
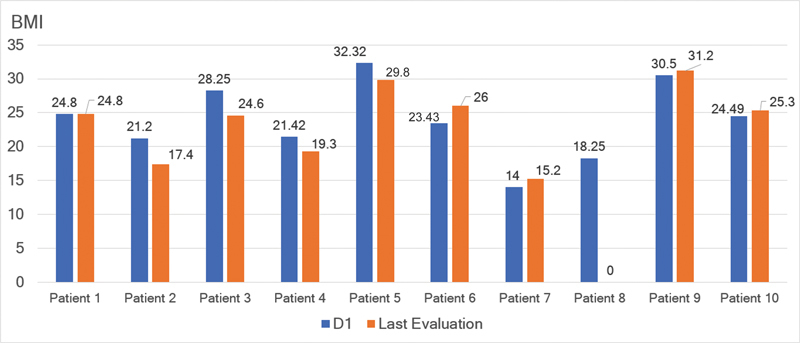
Results: Ten ATTRv-PN patients with Val30Met mutation were included. The mean disease duration on D1 was of 3 years, and the mean age of the patients was of 46.8 years. During an additional 18-month follow up, neurological function, based on the Neuropathy Impairment Score and the Polyneuropathy Disability Score, functionality aspects (Karnofsky Performance Status), and nutritional and cardiac aspects were maintained. No new safety signs have been noted.
Conclusion: The treatment with inotersen was effective and well tolerated for the average of 67 months and up to 76 months. Our results are consistent with those of larger phase-III trials.
Antecedentes: Amiloidose hereditária por transtirretina (ATTRv) é uma doença hereditária, progressiva e fatal ainda largamente subdiagnosticada. Mutações no gene transtirretina (TTR) promovem desestabilização, desdobramento, agregação e depósito da proteína TTR em tecidos do corpo, o que faz da ATTRv uma doença de fenótipo clínico heterogêneo.
Objetivo: Descrever a eficácia e segurança da terapia com inotersena no longo prazo em pacientes com neuropatia periférica ATTRv (ATTRv-PN). MéTODOS: Pacientes que completaram o estudo pivotal NEURO-TTR e o estudo de extensão aberta NEURO-TTR OLE migraram para este estudo e foram acompanhados por no mínimo 18 meses adicionais, em média por 67 meses, e por até 76 meses, desde o dia 1 da terapia com inotersena (D1–primeira dose de inotersena). A progressão da doença foi avaliada por medidas padronizadas.
Resultados: Dez pacientes com ATTRv-PN com mutação Val30Met foram incluídos. A duração média da doença no D1 era de 3 anos, e a média de idade dos pacientes era de 46,8 anos. Durante o período de acompanhamento adicional de 18 meses, a função neurológica, baseada no Neuropathy Impairment Score e no Polyneuropathy Disability Score, os aspectos de funcionalidade (Karnofsky Performance Status), nutricional e cardíacos estavam mantidos. Não se observou nenhum novo sinal de segurança. CONCLUSãO: O tratamento com inotersena foi eficaz e bem tolerado por 67 meses em média, e por até 76 meses. Nossos resultados são consistentes com os de estudos maiores de fase III.
The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution 4.0 International License, permitting copying and reproduction so long as the original work is given appropriate credit (https://creativecommons.org/licenses/by/4.0/).
Conflict of interest statement
MWC received honoraria from NHI, Prothena, FoldRx, Ionis Pharmaceuticals, Akcea Therapeutics, Pfizer, Alnylan Pharmaceuticals, PTC Therapeutics, SOBI, and Genzyme for travel expenses, as a consultant member and as principal investigator in clinical trials. No potential competing interest was reported by the other authors.
Figures
Similar articles
-
Inotersen for the treatment of adults with polyneuropathy caused by hereditary transthyretin-mediated amyloidosis.Expert Rev Clin Pharmacol. 2019 Aug;12(8):701-711. doi: 10.1080/17512433.2019.1635008. Epub 2019 Jul 3. Expert Rev Clin Pharmacol. 2019. PMID: 31268366 Review.
-
Early Data on Long-term Impact of Inotersen on Quality-of-Life in Patients with Hereditary Transthyretin Amyloidosis Polyneuropathy: Open-Label Extension of NEURO-TTR.Neurol Ther. 2021 Dec;10(2):865-886. doi: 10.1007/s40120-021-00268-x. Epub 2021 Aug 5. Neurol Ther. 2021. PMID: 34355354 Free PMC article.
-
Early data on long-term efficacy and safety of inotersen in patients with hereditary transthyretin amyloidosis: a 2-year update from the open-label extension of the NEURO-TTR trial.Eur J Neurol. 2020 Aug;27(8):1374-1381. doi: 10.1111/ene.14285. Epub 2020 May 29. Eur J Neurol. 2020. PMID: 32343462 Free PMC article. Clinical Trial.
-
Factors associated with increased health-related quality-of-life benefits in hereditary transthyretin amyloidosis polyneuropathy patients treated with inotersen.Muscle Nerve. 2022 Sep;66(3):319-328. doi: 10.1002/mus.27668. Epub 2022 Jul 15. Muscle Nerve. 2022. PMID: 35766224 Clinical Trial.
-
[Gene therapy options for hereditary transthyretin-related amyloidosis].Nervenarzt. 2022 Jun;93(6):557-565. doi: 10.1007/s00115-022-01288-0. Epub 2022 Apr 13. Nervenarzt. 2022. PMID: 35419654 Review. German.
References
-
- Pinto M V, Barreira A A, Bulle A S et al.Brazilian consensus for diagnosis, management and treatment of transthyretin familial amyloid polyneuropathy. Arq Neuropsiquiatr. 2018;76(09):609–621. - PubMed
-
- Dohrn M F, Ihne S, Hegenbart U et al.Targeting transthyretin - Mechanism-based treatment approaches and future perspectives in hereditary amyloidosis. J Neurochem. 2021;156(06):802–818. - PubMed
-
- Gertz M A, Scheinberg M, Waddington-Cruz M et al.Inotersen for the treatment of adults with polyneuropathy caused by hereditary transthyretin-mediated amyloidosis. Expert Rev Clin Pharmacol. 2019;12(08):701–711. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous