

Repressive H3K27me3 drives hyperglycemia-induced oxidative and inflammatory transcriptional programs in human endothelium
- PMID: 38580969
- PMCID: PMC10998410
- DOI: 10.1186/s12933-024-02196-0
Repressive H3K27me3 drives hyperglycemia-induced oxidative and inflammatory transcriptional programs in human endothelium
Abstract
Background: Histone modifications play a critical role in chromatin remodelling and regulate gene expression in health and disease. Histone methyltransferases EZH1, EZH2, and demethylases UTX, JMJD3, and UTY catalyse trimethylation of lysine 27 on histone H3 (H3K27me3). This study was designed to investigate whether H3K27me3 triggers hyperglycemia-induced oxidative and inflammatory transcriptional programs in the endothelium.
Methods: We studied human aortic endothelial cells exposed to high glucose (HAEC) or isolated from individuals with diabetes (D-HAEC). RT-qPCR, immunoblotting, chromatin immunoprecipitation (ChIP-qPCR), and confocal microscopy were performed to investigate the role of H3K27me3. We determined superoxide anion (O2-) production by ESR spectroscopy, NF-κB binding activity, and monocyte adhesion. Silencing/overexpression and pharmacological inhibition of chromatin modifying enzymes were used to modulate H3K27me3 levels. Furthermore, isometric tension studies and immunohistochemistry were performed in aorta from wild-type and db/db mice.
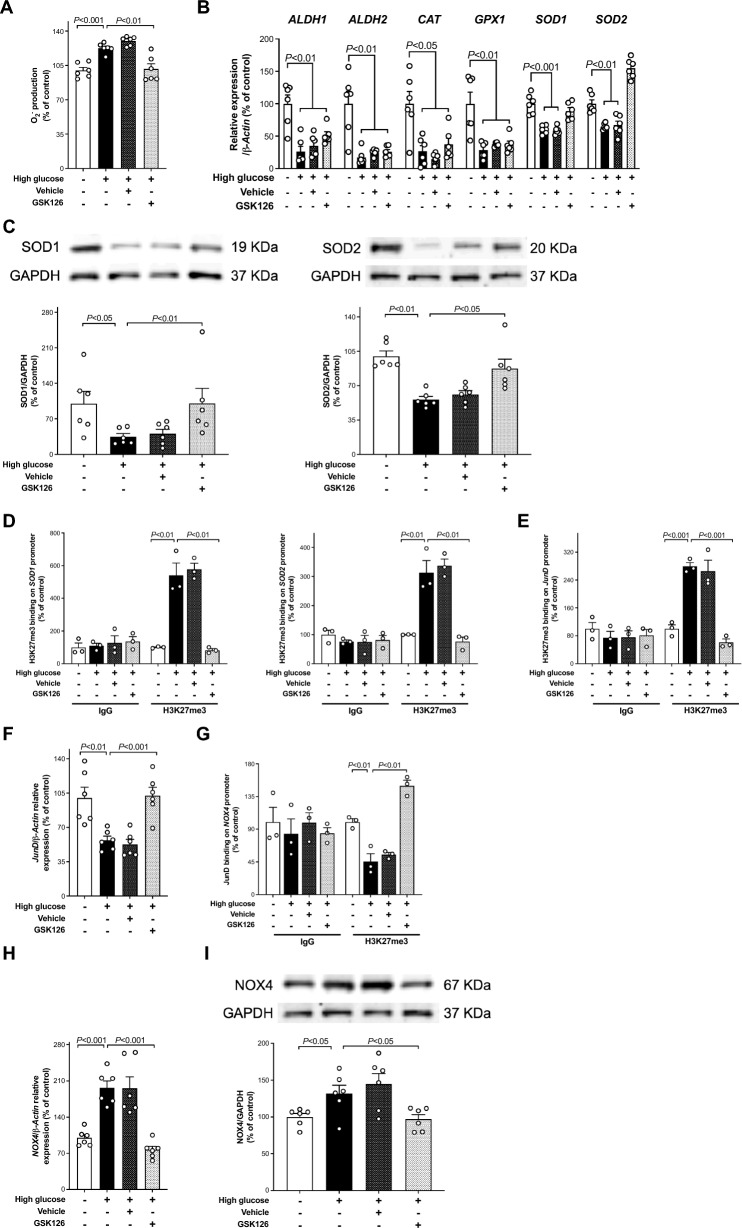
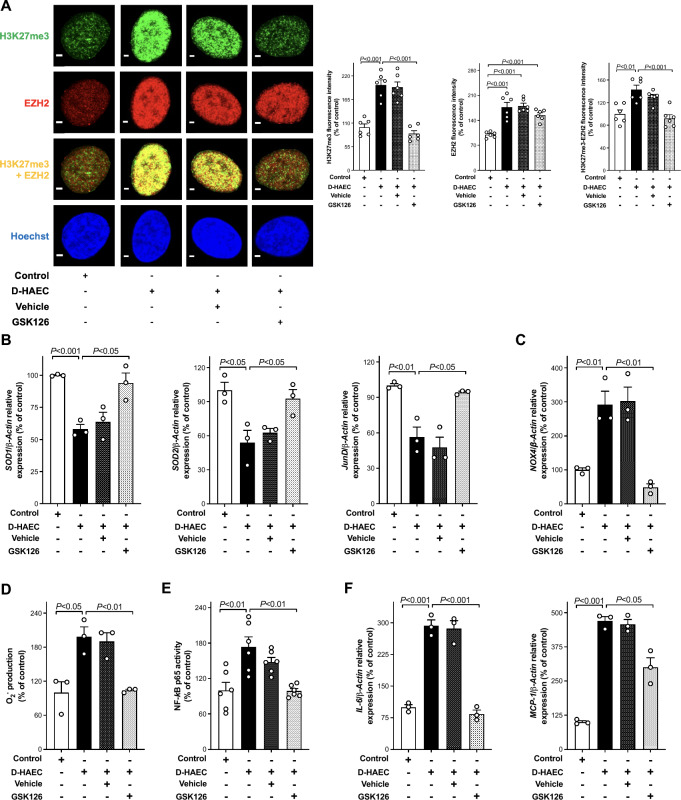
Results: Incubation of HAEC to high glucose showed that upregulation of EZH2 coupled to reduced demethylase UTX and JMJD3 was responsible for the increased H3K27me3. ChIP-qPCR revealed that repressive H3K27me3 binding to superoxide dismutase and transcription factor JunD promoters is involved in glucose-induced O2- generation. Indeed, loss of JunD transcriptional inhibition favours NOX4 expression. Furthermore, H3K27me3-driven oxidative stress increased NF-κB p65 activity and downstream inflammatory genes. Interestingly, EZH2 inhibitor GSK126 rescued these endothelial derangements by reducing H3K27me3. We also found that H3K27me3 epigenetic signature alters transcriptional programs in D-HAEC and aortas from db/db mice.
Conclusions: EZH2-mediated H3K27me3 represents a key epigenetic driver of hyperglycemia-induced endothelial dysfunction. Targeting EZH2 may attenuate oxidative stress and inflammation and, hence, prevent vascular disease in diabetes.
Keywords: Chromatin-modifying drugs; Diabetes; EZH2 inhibitor GSK126; Endothelial cells; Epigenetics; Inflammation; Oxidative stress.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that no conflict of interest exists.
Figures



References
-
- Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2019;157:107843. doi: 10.1016/j.diabres.2019.107843. - DOI - PubMed
-
- Alexander Y, Osto E, Schmidt-Trucksäss A, Shechter M, Trifunovic D, Duncker DJ, et al. Endothelial function in cardiovascular medicine: a consensus paper of the European Society of Cardiology Working Groups on Atherosclerosis and Vascular Biology, Aorta and Peripheral Vascular Diseases, Coronary Pathophysiology and Microcirculation, and Thrombosis. Cardiovasc Res. 2021;117:29–42. doi: 10.1093/cvr/cvaa085. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
