

Burden of Mendelian disorders in a large Middle Eastern biobank
- PMID: 38584274
- PMCID: PMC11000384
- DOI: 10.1186/s13073-024-01307-6
Burden of Mendelian disorders in a large Middle Eastern biobank
Abstract
Background: Genome sequencing of large biobanks from under-represented ancestries provides a valuable resource for the interrogation of Mendelian disease burden at world population level, complementing small-scale familial studies.
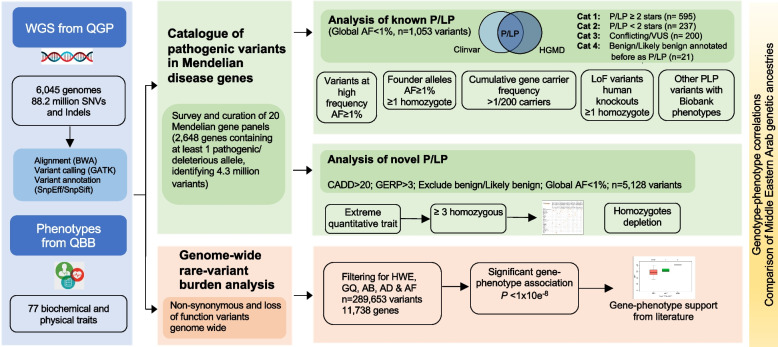
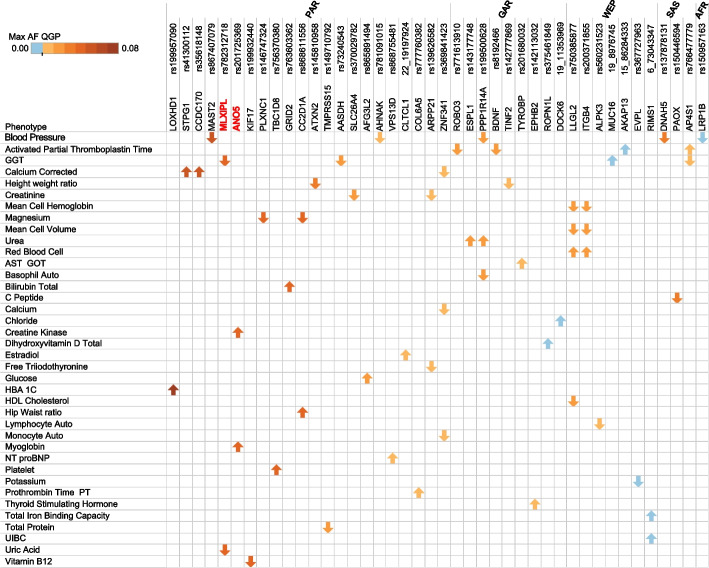
Methods: Here, we interrogate 6045 whole genomes from Qatar-a Middle Eastern population with high consanguinity and understudied mutational burden-enrolled at the national Biobank and phenotyped for 58 clinically-relevant quantitative traits. We examine a curated set of 2648 Mendelian genes from 20 panels, annotating known and novel pathogenic variants and assessing their penetrance and impact on the measured traits.
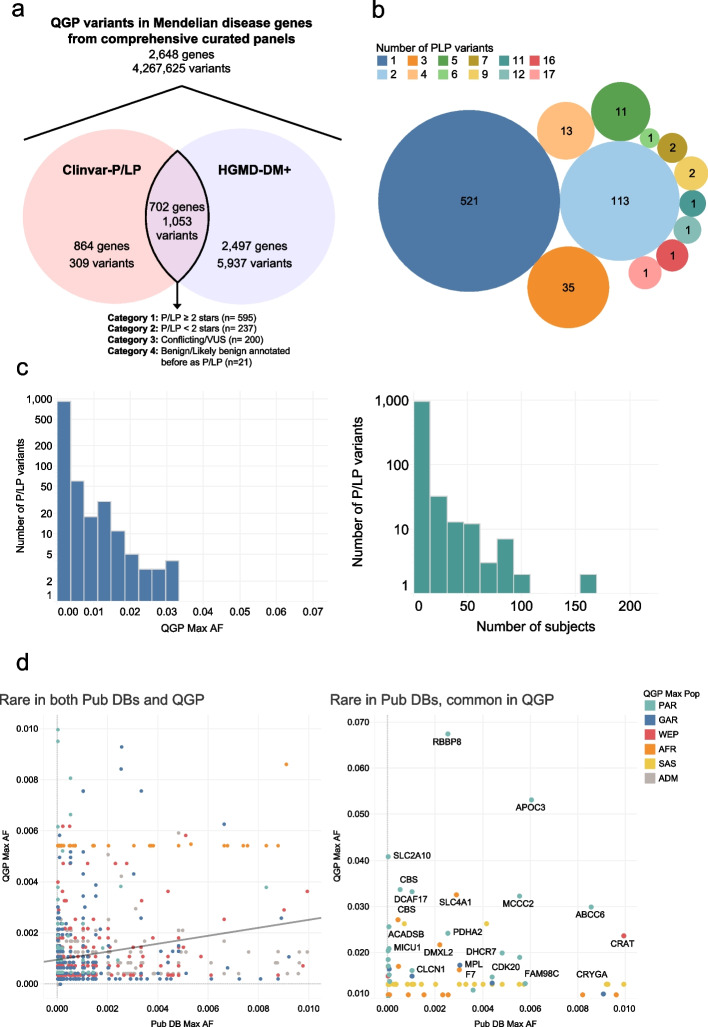
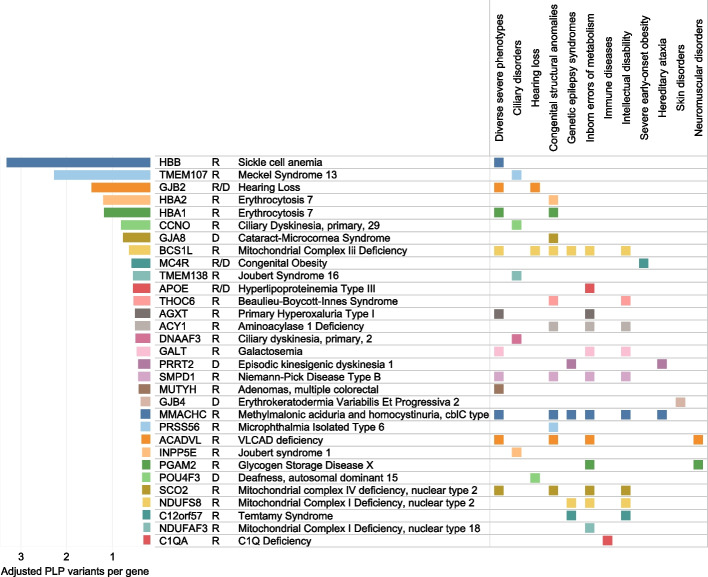
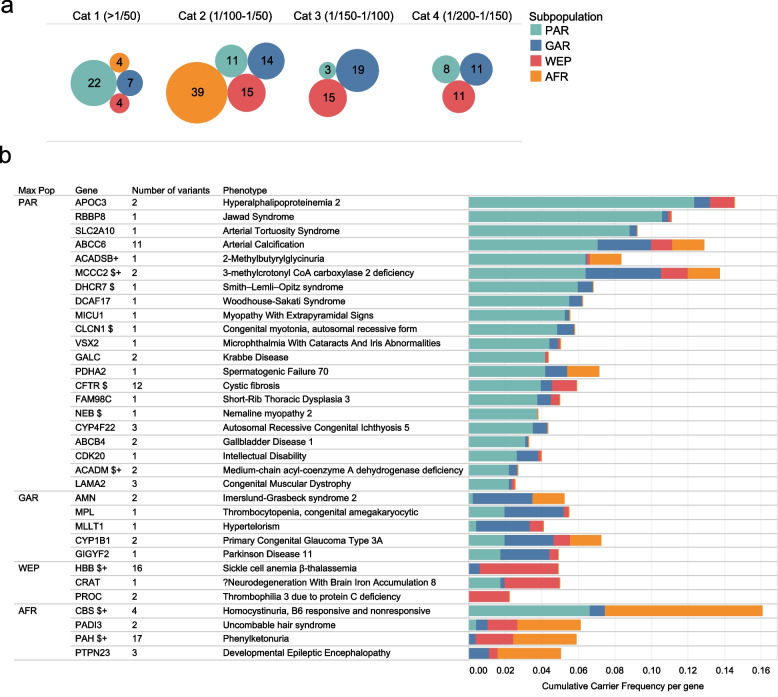
Results: We find that 62.5% of participants are carriers of at least 1 known pathogenic variant relating to recessive conditions, with homozygosity observed in 1 in 150 subjects (0.6%) for which Peninsular Arabs are particularly enriched versus other ancestries (5.8-fold). On average, 52.3 loss-of-function variants were found per genome, 6.5 of which affect a known Mendelian gene. Several variants annotated in ClinVar/HGMD as pathogenic appeared at intermediate frequencies in this cohort (1-3%), highlighting Arab founder effect, while others have exceedingly high frequencies (> 5%) prompting reconsideration as benign. Furthermore, cumulative gene burden analysis revealed 56 genes having gene carrier frequency > 1/50, including 5 ACMG Tier 3 panel genes which would be candidates for adding to newborn screening in the country. Additionally, leveraging 58 biobank traits, we systematically assess the impact of novel/rare variants on phenotypes and discover 39 candidate large-effect variants associating with extreme quantitative traits. Furthermore, through rare variant burden testing, we discover 13 genes with high mutational load, including 5 with impact on traits relevant to disease conditions, including metabolic disorder and type 2 diabetes, consistent with the high prevalence of these conditions in the region.
Conclusions: This study on the first phase of the growing Qatar Genome Program cohort provides a comprehensive resource from a Middle Eastern population to understand the global mutational burden in Mendelian genes and their impact on traits in seemingly healthy individuals in high consanguinity settings.
Keywords: Arab population; Biobank; Consanguinity; Genome sequencing; Mendelian disorders; Middle East; Pathogenic variants; Qatar; Rare genetic disease.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Fakhro KA, Robay A, Rodrigues-Flores JL, Mezey JG, Al-Shakaki AA, Chidiac O, et al. Point of care exome sequencing reveals allelic and phenotypic heterogeneity underlying Mendelian disease in Qatar. Hum Mol Genet. 2019;28(23):3970–81. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
