

Engineering allorejection-resistant CAR-NKT cells from hematopoietic stem cells for off-the-shelf cancer immunotherapy
- PMID: 38584391
- PMCID: PMC11184334
- DOI: 10.1016/j.ymthe.2024.04.005
Engineering allorejection-resistant CAR-NKT cells from hematopoietic stem cells for off-the-shelf cancer immunotherapy
Abstract
The clinical potential of current FDA-approved chimeric antigen receptor (CAR)-engineered T (CAR-T) cell therapy is encumbered by its autologous nature, which presents notable challenges related to manufacturing complexities, heightened costs, and limitations in patient selection. Therefore, there is a growing demand for off-the-shelf universal cell therapies. In this study, we have generated universal CAR-engineered NKT (UCAR-NKT) cells by integrating iNKT TCR engineering and HLA gene editing on hematopoietic stem cells (HSCs), along with an ex vivo, feeder-free HSC differentiation culture. The UCAR-NKT cells are produced with high yield, purity, and robustness, and they display a stable HLA-ablated phenotype that enables resistance to host cell-mediated allorejection. These UCAR-NKT cells exhibit potent antitumor efficacy to blood cancers and solid tumors, both in vitro and in vivo, employing a multifaceted array of tumor-targeting mechanisms. These cells are further capable of altering the tumor microenvironment by selectively depleting immunosuppressive tumor-associated macrophages and myeloid-derived suppressor cells. In addition, UCAR-NKT cells demonstrate a favorable safety profile with low risks of graft-versus-host disease and cytokine release syndrome. Collectively, these preclinical studies underscore the feasibility and significant therapeutic potential of UCAR-NKT cell products and lay a foundation for their translational and clinical development.
Keywords: CRISPR-Cas9 gene editing; T cell receptor gene engineering; allogeneic cell therapy; allorejection resistance; chimeric antigen receptor engineering; hematopoietic stem cell engineering; invariant natural killer T cell; off-the-shelf cancer immunotherapy; tumor microenvironment; universal CAR-engineered NKT cell.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests Y.-R.L., Y. Zhou, J.Y., D.L., Z.L., Y.J.K., P.W., and L.Y. are inventors on patents relating to this study filed by UCLA. Y. Zhou is currently an employee of Amberstone Biosciences. J.Y. is currently an employee of Appia Bio. Z.L. is currently an employee of Allogene. Y.J.K. is currently an employee of Nkarta. P.W. is a co-founder, stockholder, and advisory board member of Simnova Bio, TCRCure Biopharma, Appia Bio, and is a scientific advisor to Grit Biotechnolgoy. L.Y. is a scientific advisor to AlzChem and Amberstone Biosciences, and a co-founder, stockholder, and advisory board member of Appia Bio. Appia Bio licensed some patents relating to this study from UCLA. None of the declared companies contributed to or directed any of the research reported in this article. The remaining authors declare no competing interests.
Figures


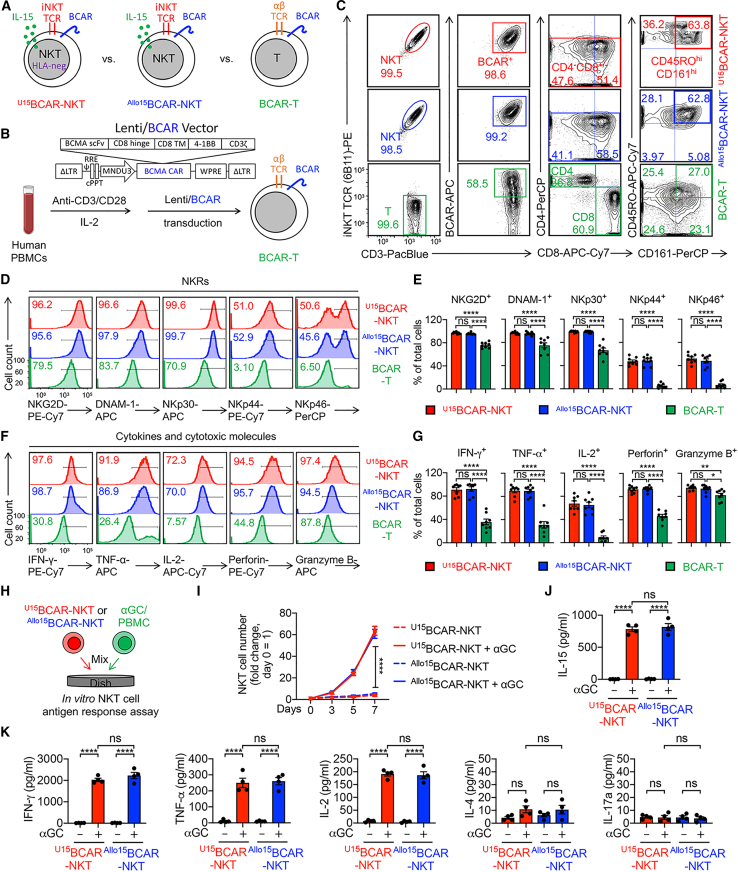

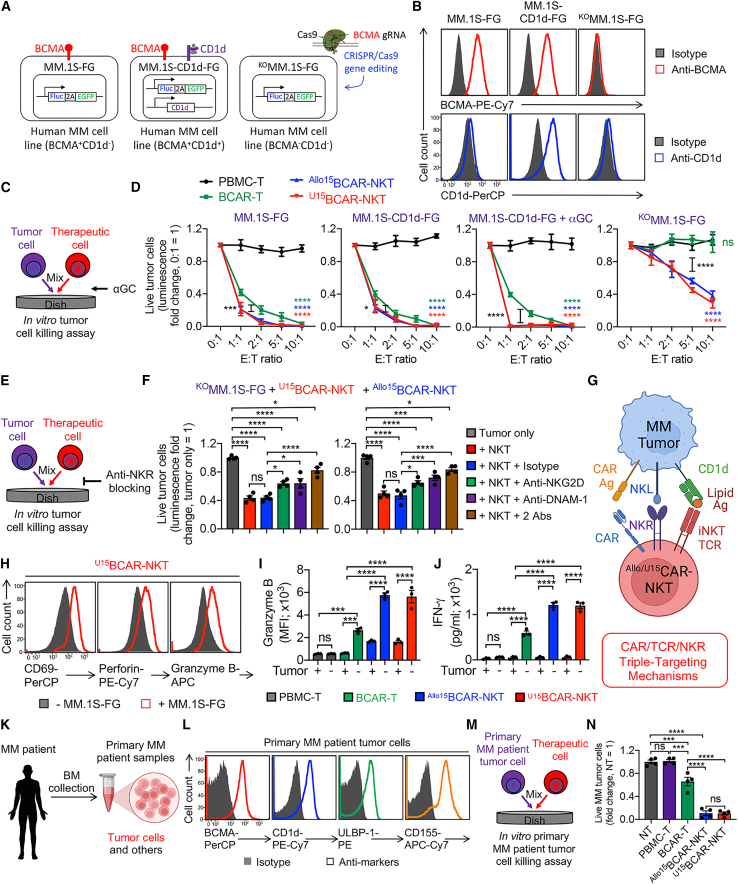



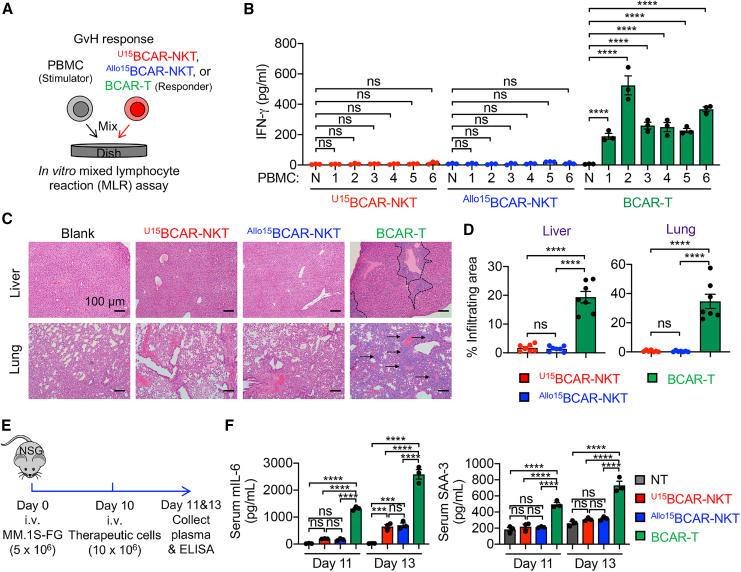
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
