

Triple-A Syndrome in Morocco: Founder Effect, Age Estimation of the AAAS c.1331+1G>A Variant, and Implications for Genetic Diagnosis
- PMID: 38585542
- PMCID: PMC10996341
- DOI: 10.1159/000533894
Triple-A Syndrome in Morocco: Founder Effect, Age Estimation of the AAAS c.1331+1G>A Variant, and Implications for Genetic Diagnosis
Abstract
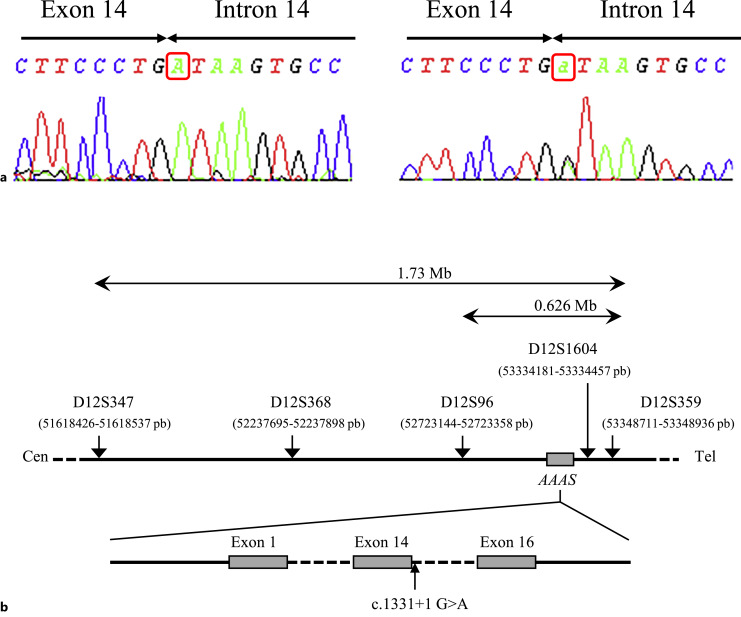
Introduction: Triple-A syndrome (Triple-A) is an autosomal recessive disorder characterized by alacrimia, achalasia, and adrenal insufficiency. Several variants on the AAAS gene have been described, and some variants are clustered in particular geographical areas, such as the c.1331+1G>A variant which is very frequent in North Africa. Here, we describe the genetic features of Triple-A in a series of unrelated families from Morocco.
Methods: Screening for the AAAS c.1331+1G>A variant was performed by direct sequencing or by PCR-RFLP. Haplotype analysis using Single Tandem Repeat (STR) markers flanking AAAS gene was performed in order to evaluate the founder effect and estimate the age of the c.1331+1G>A variant.
Results: Seven unrelated families with ten individuals clinically diagnosed with Triple-A were evaluated for sequence variations in the AAAS gene. The median age at diagnosis was 3 years, with a range between 2 and 11 years. Molecular analysis revealed that all patients were homozygous for the c.1331+1G>A variant. This variant was not found in 200 healthy controls, indicating that carriers are very rare in the general Moroccan population. Subsequently, STR marker analysis revealed a founder effect and that the most recent common ancestor of Triple-A patients in Morocco would have lived 125 years ago.
Conclusion: This is the largest series of Triple-A in Morocco. The same AAAS c.1331+1G>A variant was found in all patients, suggesting a founder effect in Morocco which was subsequently confirmed by microsatellite marker analysis. Therefore, this variant should be systematically investigated to diagnose Triple-A in Morocco.
Keywords: AAAS gene; Allgrove syndrome; BRO Biobank; Carrier frequency; Founder effect; Triple-A syndrome; c.1331+1G > A variant.
© 2023 S. Karger AG, Basel.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

References
-
- Achargui M, Azhrai R, Harrar Y, Mabrouki F, Chariba S, Maadane A, et al. . Alacrimia revealing triple A syndrome: a case report. Asian J Ophthalmol Case Rep. 2023;6:24–8.
-
- Amrani R, Es-seddiki A, Ayyad A, Messaoudi S, Tazi N. Syndrome d’Allgrove découvert sur une anémie ferriprive chez un enfant de 3 ans. Pan Afr Med J. 2014;19. 10.11604/pamj.2014.19.218.5511. - DOI
LinkOut - more resources
Full Text Sources
