

This is a preprint.
Deep learning to decode sites of RNA translation in normal and cancerous tissues
- PMID: 38585907
- PMCID: PMC10996544
- DOI: 10.1101/2024.03.21.586110
Deep learning to decode sites of RNA translation in normal and cancerous tissues
Update in
-
Deep learning to decode sites of RNA translation in normal and cancerous tissues.Nat Commun. 2025 Feb 2;16(1):1275. doi: 10.1038/s41467-025-56543-0. Nat Commun. 2025. PMID: 39894899 Free PMC article.
Abstract
The biological process of RNA translation is fundamental to cellular life and has wide-ranging implications for human disease. Yet, accurately delineating the variation in RNA translation represents a significant challenge. Here, we develop RiboTIE, a transformer model-based approach to map global RNA translation. We find that RiboTIE offers unparalleled precision and sensitivity for ribosome profiling data. Application of RiboTIE to normal brain and medulloblastoma cancer samples enables high-resolution insights into disease regulation of RNA translation.
Keywords: RNA translation; Ribo-seq; cancer; medulloblastoma; non-canonical open reading frames.
Conflict of interest statement
Declaraon of Interests G.M. is an employee of OHMX Bio. Z.M. and R.G. are employees of Novo Nordisk Ltd. J.R.P. reports receiving honoraria from Novartis Biosciences.
Figures
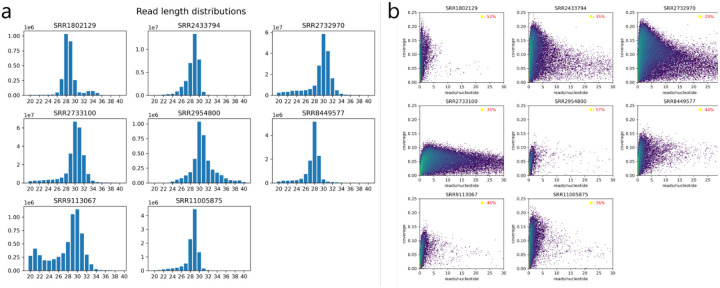
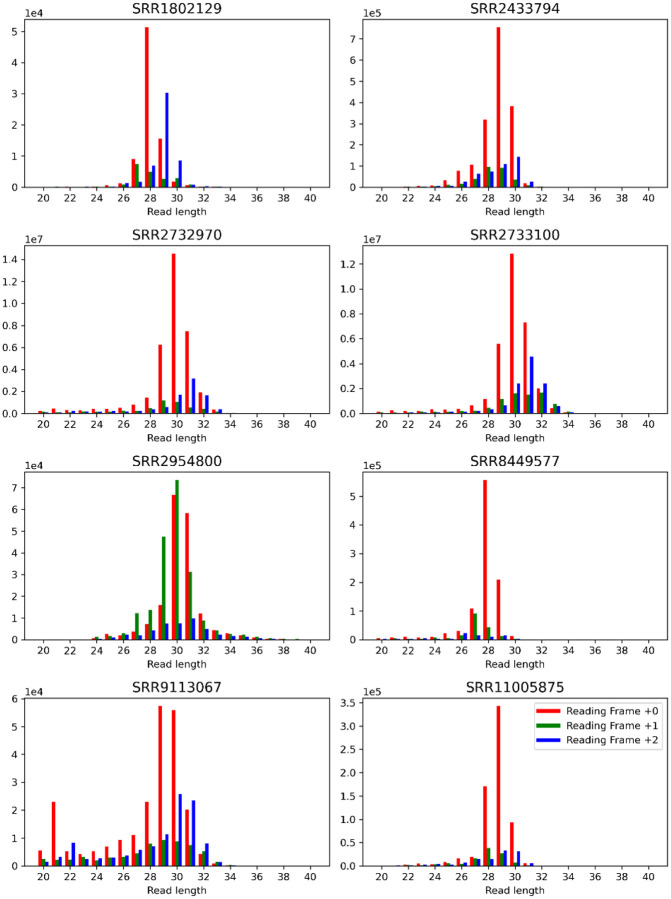
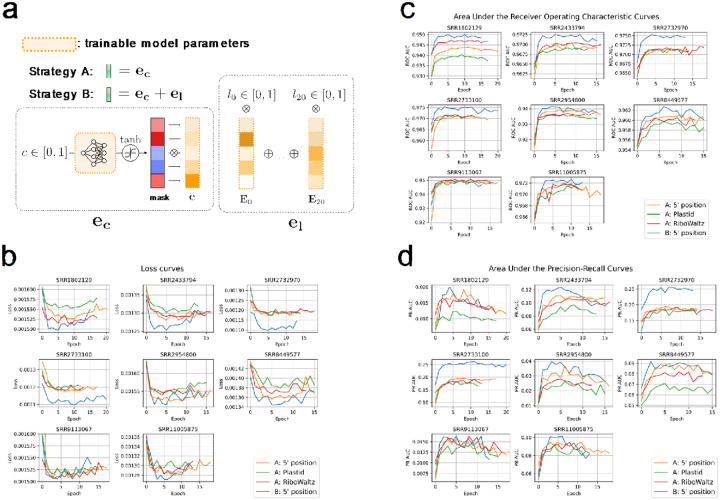
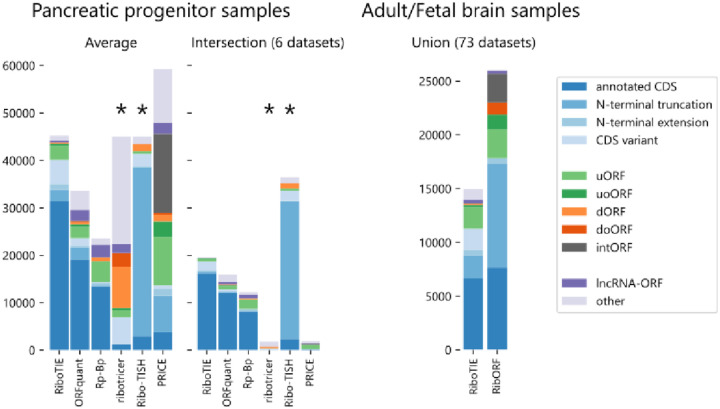
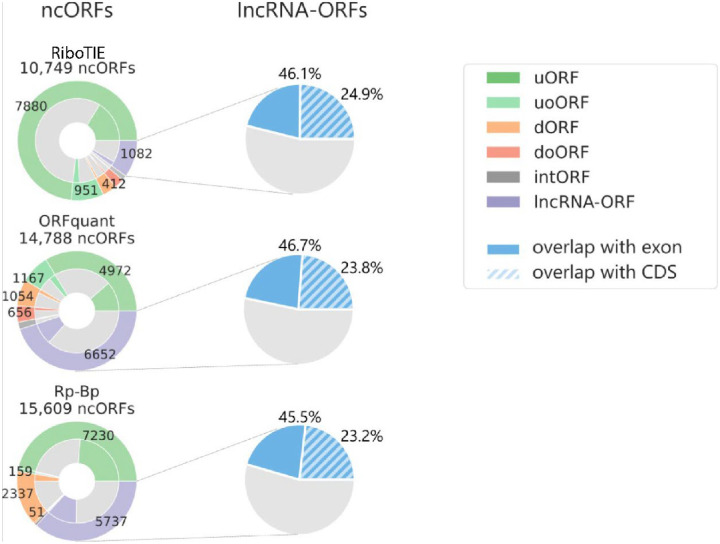
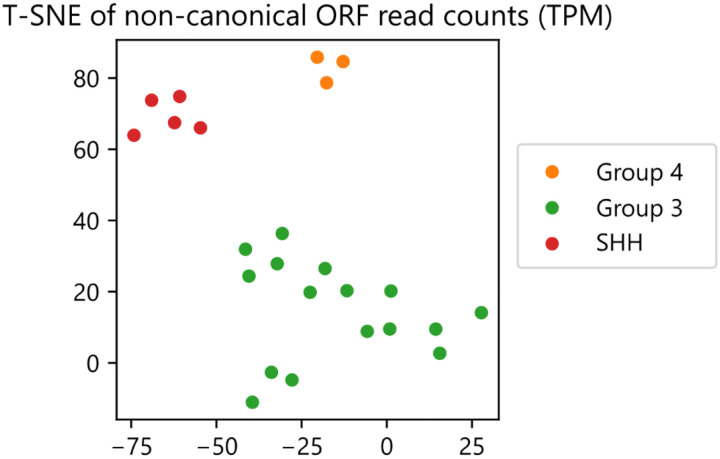
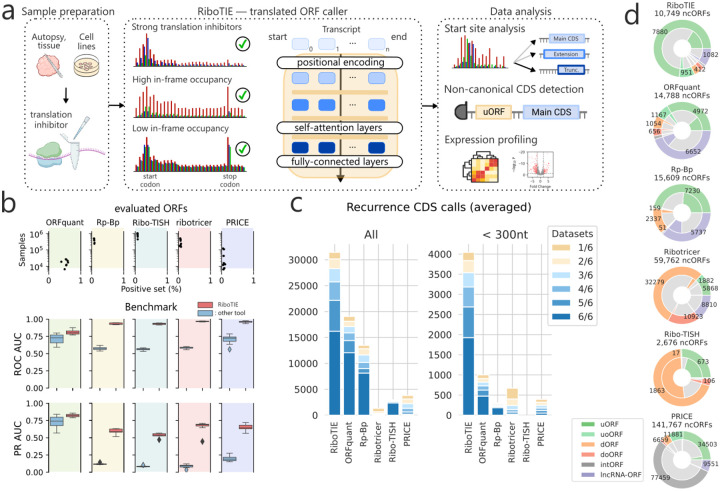
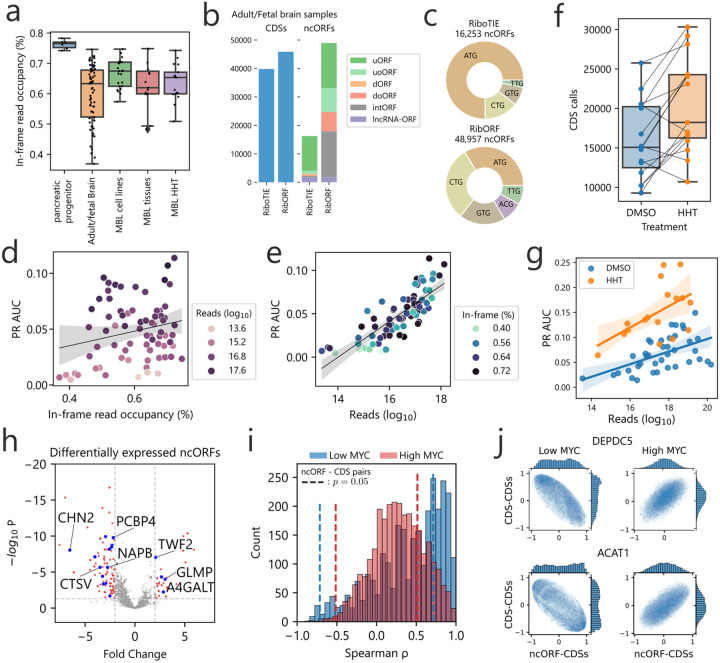
References
-
- Prensner J. R., Abelin J.G., Kok L.W., Clauser K.R., Mudge J.M., Ruiz-Orera J., Bassini-Sternberg M., Deutsch E.W., Moritz R.L., van Heesch S. What can Ribo-seq, immunopeptidomics, and proteomics tell us about the non-canonical proteome? Mol Cell Proteomics 22 (2023). 10.1016/j.mcpro.2023.100631. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources